All organisms must duplicate their DNA with extraordinary accuracy before each cell division. In this section, we explore how an elaborate “replication machine” achieves this accuracy, while duplicating DNA at rates as high as 1000 nucleotides per second.
Base-Pairing Underlies DNA Replication and DNA Repair
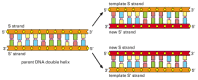
As discussed briefly in Chapter 1, DNA templating is the process in which the nucleotide sequence of a DNA strand (or selected portions of a DNA strand) is copied by complementary base-pairing (A with T, and G with C) into a complementary DNA sequence (). This process entails the recognition of each nucleotide in the DNA template strand by a free (unpolymerized) complementary nucleotide, and it requires that the two strands of the DNA helix be separated. This separation allows the hydrogen-bond donor and acceptor groups on each DNA base to become exposed for base-pairing with the appropriate incoming free nucleotide, aligning it for its enzyme-catalyzed polymerization into a new DNA chain.
The DNA double helix acts as a template for its own duplication. Because the nucleotide A will successfully pair only with T, and G only with C, each strand of DNA can serve as a template to specify the sequence of nucleotides in its complementary strand (more...)
The first nucleotide polymerizing enzyme, DNA polymerase, was discovered in 1957. The free nucleotides that serve as substrates for this enzyme were found to be deoxyribonucleoside triphosphates, and their polymerization into DNA required a single-stranded DNA template. The stepwise mechanism of this reaction is illustrated in and .
The chemistry of DNA synthesis. The addition of a deoxyribonucleotide to the 3′ end of a polynucleotide chain (the primer strand) is the fundamental reaction by which DNA is synthesized. As shown, base-pairing between an incoming deoxyribonucleoside (more...)
DNA synthesis catalyzed by DNA polymerase. (A) As indicated, DNA polymerase catalyzes the stepwise addition of a deoxyribonucleotide to the 3′-OH end of a polynucleotide chain, the primer strand, that is paired to a second template strand. The (more...)
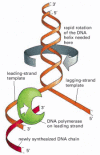
The DNA Replication Fork Is Asymmetrical
During DNA replication inside a cell, each of the two old DNA strands serves as a template for the formation of an entire new strand. Because each of the two daughters of a dividing cell inherits a new DNA double helix containing one old and one new strand (), the DNA double helix is said to be replicated “semiconservatively” by DNA polymerase. How is this feat accomplished?
The semiconservative nature of DNA replication. In a round of replication, each of the two strands of DNA is used as a template for the formation of a complementary DNA strand. The original strands therefore remain intact through many cell generations. (more...)
Analyses carried out in the early 1960s on whole replicating chromosomes revealed a localized region of replication that moves progressively along the parental DNA double helix. Because of its Y-shaped structure, this active region is called a replication fork (). At a replication fork, the DNA of both new daughter strands is synthesized by a multienzyme complex that contains the DNA polymerase.
Two replication forks moving in opposite directions on a circular chromosome. An active zone of DNA replication moves progressively along a replicating DNA molecule, creating a Y-shaped DNA structure known as a replication fork: the two arms of each Y (more...)
Initially, the simplest mechanism of DNA replication seemed to be the continuous growth of both new strands, nucleotide by nucleotide, at the replication fork as it moves from one end of a DNA molecule to the other. But because of the antiparallel orientation of the two DNA strands in the DNA double helix (see ), this mechanism would require one daughter strand to polymerize in the 5′-to-3′ direction and the other in the 3′-to-5′ direction. Such a replication fork would require two different DNA polymerase enzymes. One would polymerize in the 5′-to-3′ direction, where each incoming deoxyribonucleoside triphosphate carried the triphosphate activation needed for its own addition. The other would move in the 3′-to-5′ direction and work by so-called “head growth,” in which the end of the growing DNA chain carried the triphosphate activation required for the addition of each subsequent nucleotide (). Although head-growth polymerization occurs elsewhere in biochemistry (see pp. 89–90), it does not occur in DNA synthesis; no 3′-to-5′ DNA polymerase has ever been found.
An incorrect model for DNA replication. Although it might seem to be the simplest possible model for DNA replication, the mechanism illustrated here is not the one that cells use. In this scheme, both daughter DNA strands would grow continuously, using (more...)
How, then, is overall 3′-to-5′ DNA chain growth achieved? The answer was first suggested by the results of experiments in the late 1960s. Researchers added highly radioactive 3H-thymidine to dividing bacteria for a few seconds, so that only the most recently replicated DNA—that just behind the replication fork—became radiolabeled. This experiment revealed the transient existence of pieces of DNA that were 1000–2000 nucleotides long, now commonly known as Okazaki fragments, at the growing replication fork. (Similar replication intermediates were later found in eucaryotes, where they are only 100–200 nucleotides long.) The Okazaki fragments were shown to be polymerized only in the 5′-to-3′chain direction and to be joined together after their synthesis to create long DNA chains.
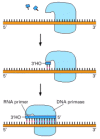
A replication fork therefore has an asymmetric structure (). The DNA daughter strand that is synthesized continuously is known as the leading strand. Its synthesis slightly precedes the synthesis of the daughter strand that is synthesized discontinuously, known as the lagging strand. For the lagging strand, the direction of nucleotide polymerization is opposite to the overall direction of DNA chain growth. Lagging-strand DNA synthesis is delayed because it must wait for the leading strand to expose the template strand on which each Okazaki fragment is synthesized. The synthesis of the lagging strand by a discontinuous “backstitching” mechanism means that only the 5′-to-3′ type of DNA polymerase is needed for DNA replication.
The structure of a DNA replication fork. Because both daughter DNA strands are polymerized in the 5′-to-3′ direction, the DNA synthesized on the lagging strand must be made initially as a series of short DNA molecules, called Okazaki fragments. (more...)
The High Fidelity of DNA Replication Requires Several Proofreading Mechanisms
As discussed at the beginning of this chapter, the fidelity of copying DNA during replication is such that only about 1 mistake is made for every 109 nucleotides copied. This fidelity is much higher than one would expect, on the basis of the accuracy of complementary base-pairing. The standard complementary base pairs (see ) are not the only ones possible. For example, with small changes in helix geometry, two hydrogen bonds can form between G and T in DNA. In addition, rare tautomeric forms of the four DNA bases occur transiently in ratios of 1 part to 104 or 105. These forms mispair without a change in helix geometry: the rare tautomeric form of C pairs with A instead of G, for example.
If the DNA polymerase did nothing special when a mispairing occurred between an incoming deoxyribonucleoside triphosphate and the DNA template, the wrong nucleotide would often be incorporated into the new DNA chain, producing frequent mutations. The high fidelity of DNA replication, however, depends not only on complementary base-pairing but also on several “proofreading” mechanisms that act sequentially to correct any initial mispairing that might have occurred.
The first proofreading step is carried out by the DNA polymerase, and it occurs just before a new nucleotide is added to the growing chain. Our knowledge of this mechanism comes from studies of several different DNA polymerases, including one produced by a bacterial virus, T7, that replicates inside E. coli. The correct nucleotide has a higher affinity for the moving polymerase than does the incorrect nucleotide, because only the correct nucleotide can correctly base-pair with the template. Moreover, after nucleotide binding, but before the nucleotide is covalently added to the growing chain, the enzyme must undergo a conformational change. An incorrectly bound nucleotide is more likely to dissociate during this step than the correct one. This step therefore allows the polymerase to “double-check” the exact base-pair geometry before it catalyzes the addition of the nucleotide.
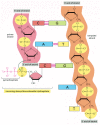
The next error-correcting reaction, known as exonucleolytic proofreading, takes place immediately after those rare instances in which an incorrect nucleotide is covalently added to the growing chain. DNA polymerase enzymes cannot begin a new polynucleotide chain by linking two nucleoside triphosphates together. Instead, they absolutely require a base-paired 3′-OH end of a primer strand on which to add further nucleotides (see ). Those DNA molecules with a mismatched (improperly base-paired) nucleotide at the 3′-OH end of the primer strand are not effective as templates because the polymerase cannot extend such a strand. DNA polymerase molecules deal with such a mismatched primer strand by means of a separate catalytic site (either in a separate subunit or in a separate domain of the polymerase molecule, depending on the polymerase). This 3′-to-5′ proofreading exonuclease clips off any unpaired residues at the primer terminus, continuing until enough nucleotides have been removed to regenerate a base-paired 3′-OH terminus that can prime DNA synthesis. In this way, DNA polymerase functions as a “self-correcting” enzyme that removes its own polymerization errors as it moves along the DNA ( and ).
Exonucleolytic proofreading by DNA polymerase during DNA replication. In this example, the mismatch is due to the incorporation of a rare, transient tautomeric form of C, indicated by an asterisk. But the same proofreading mechanism applies to any misincorporation (more...)
Editing by DNA polymerase. Outline of the structures of DNA polymerase complexed with the DNA template in the polymerizing mode (left) and the editing mode (right). The catalytic site for the exonucleolytic (E) and the polymerization (P) reactions are (more...)
The requirement for a perfectly base-paired primer terminus is essential to the self-correcting properties of the DNA polymerase. It is apparently not possible for such an enzyme to start synthesis in the complete absence of a primer without losing any of its discrimination between base-paired and unpaired growing 3′-OH termini. By contrast, the RNA polymerase enzymes involved in gene transcription do not need efficient exonucleolytic proofreading: errors in making RNA are not passed on to the next generation, and the occasional defective RNA molecule that is produced has no long-term significance. RNA polymerases are thus able to start new polynucleotide chains without a primer.
An error frequency of about 1 in 104 is found both in RNA synthesis and in the separate process of translating mRNA sequences into protein sequences. This level of mistakes is 100,000 times greater than that in DNA replication, where a series of proofreading processes makes the process remarkably accurate ().
The Three Steps That Give Rise to High-Fidelity DNA Synthesis.
Only DNA Replication in the 5′-to-3′ Direction Allows Efficient Error Correction
The need for accuracy probably explains why DNA replication occurs only in the 5′-to-3′ direction. If there were a DNA polymerase that added deoxyribonucleoside triphosphates in the 3′-to-5′ direction, the growing 5′-chain end, rather than the incoming mononucleotide, would carry the activating triphosphate. In this case, the mistakes in polymerization could not be simply hydrolyzed away, because the bare 5′-chain end thus created would immediately terminate DNA synthesis (). It is therefore much easier to correct a mismatched base that has just been added to the 3′ end than one that has just been added to the 5′ end of a DNA chain. Although the mechanism for DNA replication (see ) seems at first sight much more complex than the incorrect mechanism depicted earlier in , it is much more accurate because all DNA synthesis occurs in the 5′-to-3′ direction.
An explanation for the 5′-to-3′ direction of DNA chain growth. Growth in the 5′-to-3′ direction, shown on the right, allows the chain to continue to be elongated when a mistake in polymerization has been removed by exonucleolytic (more...)
Despite these safeguards against DNA replication errors, DNA polymerases occasionally make mistakes. However, as we shall see later, cells have yet another chance to correct these errors by a process called strand-directed mismatch repair. Before discussing this mechanism, however, we describe the other types of proteins that function at the replication fork.
A Special Nucleotide-Polymerizing Enzyme Synthesizes Short RNA Primer Molecules on the Lagging Strand
For the leading strand, a special primer is needed only at the start of replication: once a replication fork is established, the DNA polymerase is continuously presented with a base-paired chain end on which to add new nucleotides. On the lagging side of the fork, however, every time the DNA polymerase completes a short DNA Okazaki fragment (which takes a few seconds), it must start synthesizing a completely new fragment at a site further along the template strand (see ). A special mechanism is used to produce the base-paired primer strand required by this DNA polymerase molecule. The mechanism involves an enzyme called DNA primase, which uses ribonucleoside triphosphates to synthesize short RNA primers on the lagging strand (). In eucaryotes, these primers are about 10 nucleotides long and are made at intervals of 100–200 nucleotides on the lagging strand.
RNA primer synthesis. A schematic view of the reaction catalyzed by DNA primase, the enzyme that synthesizes the short RNA primers made on the lagging strand using DNA as a template. Unlike DNA polymerase, this enzyme can start a new polynucleotide chain (more...)
The chemical structure of RNA was introduced in Chapter 1 and described in detail in Chapter 6. Here, we note only that RNA is very similar in structure to DNA. A strand of RNA can form base pairs with a strand of DNA, generating a DNA/RNA hybrid double helix if the two nucleotide sequences are complementary. The synthesis of RNA primers is thus guided by the same templating principle used for DNA synthesis (see and ).
Because an RNA primer contains a properly base-paired nucleotide with a 3′-OH group at one end, it can be elongated by the DNA polymerase at this end to begin an Okazaki fragment. The synthesis of each Okazaki fragment ends when this DNA polymerase runs into the RNA primer attached to the 5′ end of the previous fragment. To produce a continuous DNA chain from the many DNA fragments made on the lagging strand, a special DNA repair system acts quickly to erase the old RNA primer and replace it with DNA. An enzyme called DNA ligase then joins the 3′ end of the new DNA fragment to the 5′ end of the previous one to complete the process ( and ).
The synthesis of one of the many DNA fragments on the lagging strand. In eucaryotes, RNA primers are made at intervals spaced by about 200 nucleotides on the lagging strand, and each RNA primer is approximately 10 nucleotides long. This primer is erased (more...)
The reaction catalyzed by DNA ligase. This enzyme seals a broken phosphodiester bond. As shown, DNA ligase uses a molecule of ATP to activate the 5′ end at the nick (step 1) before forming the new bond (step 2). In this way, the energetically (more...)
Why might an erasable RNA primer be preferred to a DNA primer that would not need to be erased? The argument that a self-correcting polymerase cannot start chains de novo also implies its converse: an enzyme that starts chains anew cannot be efficient at self-correction. Thus, any enzyme that primes the synthesis of Okazaki fragments will of necessity make a relatively inaccurate copy (at least 1 error in 105). Even if the copies retained in the final product constituted as little as 5% of the total genome (for example, 10 nucleotides per 200-nucleotide DNA fragment), the resulting increase in the overall mutation rate would be enormous. It therefore seems likely that the evolution of RNA rather than DNA for priming brought a powerful advantage to the cell: the ribonucleotides in the primer automatically mark these sequences as “suspect copy” to be efficiently removed and replaced.
Special Proteins Help to Open Up the DNA Double Helix in Front of the Replication Fork
For DNA synthesis to proceed, the DNA double helix must be opened up ahead of the replication fork so that the incoming deoxyribonucleoside triphosphates can form base pairs with the template strand. However, the DNA double helix is very stable under normal conditions; the base pairs are locked in place so strongly that temperatures approaching that of boiling water are required to separate the two strands in a test tube. For this reason, DNA polymerases and DNA primases can copy a DNA double helix only when the template strand has already been exposed by separating it from its complementary strand. Additional replication proteins are needed to help in opening the double helix and thus provide the appropriate single-stranded DNA template for the DNA polymerase to copy. Two types of protein contribute to this process—DNA helicases and single-strand DNA-binding proteins.
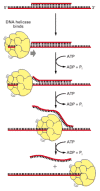
DNA helicases were first isolated as proteins that hydrolyze ATP when they are bound to single strands of DNA. As described in Chapter 3, the hydrolysis of ATP can change the shape of a protein molecule in a cyclical manner that allows the protein to perform mechanical work. DNA helicases use this principle to propel themselves rapidly along a DNA single strand. When they encounter a region of double helix, they continue to move along their strand, thereby prying apart the helix at rates of up to 1000 nucleotide pairs per second ( and ).
An assay used to test for DNA helicase enzymes. A short DNA fragment is annealed to a long DNA single strand to form a region of DNA double helix. The double helix is melted as the helicase runs along the DNA single strand, releasing the short DNA fragment (more...)
The structure of a DNA helicase. (A) A schematic diagram of the protein as a hexameric ring. (B) Schematic diagram showing a DNA replication fork and helicase to scale. (C) Detailed structure of the bacteriophage T7 replicative helicase, as determined (more...)
The unwinding of the template DNA helix at a replication fork could in principle be catalyzed by two DNA helicases acting in concert—one running along the leading strand template and one along the lagging strand template. Since the two strands have opposite polarities, these helicases would need to move in opposite directions along a DNA single strand and therefore would be different enzymes. Both types of DNA helicase exist. In the best understood replication systems, a helicase on the lagging-strand template appears to have the predominant role, for reasons that will become clear shortly.
Single-strand DNA-binding (SSB) proteins, also called helix-destabilizing proteins, bind tightly and cooperatively to exposed single-stranded DNA strands without covering the bases, which therefore remain available for templating. These proteins are unable to open a long DNA helix directly, but they aid helicases by stabilizing the unwound, single-stranded conformation. In addition, their cooperative binding coats and straightens out the regions of single-stranded DNA on the lagging-strand template, thereby preventing the formation of the short hairpin helices that readily form in single-strand DNA ( and ). These hairpin helices can impede the DNA synthesis catalyzed by DNA polymerase.
The effect of single-strand DNA-binding proteins (SSB proteins) on the structure of single-stranded DNA. Because each protein molecule prefers to bind next to a previously bound molecule, long rows of this protein form on a DNA single strand. This cooperative (more...)
The structure of the single-strand binding protein from humans bound to DNA. (A) A front view of the two DNA binding domains of RPA protein, which cover a total of eight nucleotides. Note that the DNA bases remain exposed in this protein–DNA complex. (more...)
A Moving DNA Polymerase Molecule Stays Connected to the DNA by a Sliding Ring
On their own, most DNA polymerase molecules will synthesize only a short string of nucleotides before falling off the DNA template. The tendency to dissociate quickly from a DNA molecule allows a DNA polymerase molecule that has just finished synthesizing one Okazaki fragment on the lagging strand to be recycled quickly, so as to begin the synthesis of the next Okazaki fragment on the same strand. This rapid dissociation, however, would make it difficult for the polymerase to synthesize the long DNA strands produced at a replication fork were it not for an accessory protein that functions as a regulated clamp. This clamp keeps the polymerase firmly on the DNA when it is moving, but releases it as soon as the polymerase runs into a double-stranded region of DNA ahead.
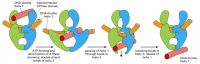
How can a clamp prevent the polymerase from dissociating without at the same time impeding the polymerase's rapid movement along the DNA molecule? The three-dimensional structure of the clamp protein, determined by x-ray diffraction, reveals that it forms a large ring around the DNA helix. One side of the ring binds to the back of the DNA polymerase, and the whole ring slides freely along the DNA as the polymerase moves. The assembly of the clamp around DNA requires ATP hydrolysis by a special protein complex, the clamp loader, which hydrolyzes ATP as it loads the clamp on to a primer-template junction ().
The regulated sliding clamp that holds DNA polymerase on the DNA. (A) The structure of the clamp protein from E. coli, as determined by x-ray crystallography, with a DNA helix added to indicate how the protein fits around DNA. (B) A similar protein is (more...)
On the leading-strand template, the moving DNA polymerase is tightly bound to the clamp, and the two remain associated for a very long time. However, on the lagging-strand template, each time the polymerase reaches the 5′ end of the preceding Okazaki fragment, the polymerase is released; this polymerase molecule then associates with a new clamp that is assembled on the RNA primer of the next Okazaki fragment ().
A cycle of loading and unloading of DNA polymerase and the clamp protein on the lagging strand. The association of the clamp loader with the lagging-strand polymerase shown here is for illustrative purposes only; in reality, the clamp loader is carried (more...)
The Proteins at a Replication Fork Cooperate to Form a Replication Machine
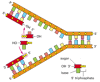
Although we have discussed DNA replication as though it were performed by a mixture of proteins all acting independently, in reality, most of the proteins are held together in a large multienzyme complex that moves rapidly along the DNA. This complex can be likened to a tiny sewing machine composed of protein parts and powered by nucleoside triphosphate hydrolyses. Although the replication complex has been most intensively studied in E. coli and several of its viruses, a very similar complex also operates in eucaryotes, as we see below.
The functions of the subunits of the replication machine are summarized in . Two DNA polymerase molecules work at the fork, one on the leading strand and one on the lagging strand. The DNA helix is opened by a DNA polymerase molecule clamped on the leading strand, acting in concert with one or more DNA helicase molecules running along the strands in front of it. Helix opening is aided by cooperatively bound molecules of single-strand DNA-binding protein. Whereas the DNA polymerase molecule on the leading strand can operate in a continuous fashion, the DNA polymerase molecule on the lagging strand must restart at short intervals, using a short RNA primer made by a DNA primase molecule.
The proteins at a bacterial DNA replication fork. The major types of proteins that act at a DNA replication fork are illustrated, showing their approximate positions on the DNA.
The efficiency of replication is greatly increased by the close association of all these protein components. In procaryotes, the primase molecule is linked directly to a DNA helicase to form a unit on the lagging strand called a primosome. Powered by the DNA helicase, the primosome moves with the fork, synthesizing RNA primers as it goes. Similarly, the DNA polymerase molecule that synthesizes DNA on the lagging strand moves in concert with the rest of the proteins, synthesizing a succession of new Okazaki fragments. To accommodate this arrangement, the lagging strand seems to be folded back in the manner shown in . This arrangement also facilitates the loading of the polymerase clamp each time that an Okazaki fragment is synthesized: the clamp loader and the lagging-strand DNA polymerase molecule are kept in place as a part of the protein machine even when they detach from the DNA. The replication proteins are thus linked together into a single large unit (total molecular weight >106 daltons) that moves rapidly along the DNA, enabling DNA to be synthesized on both sides of the replication fork in a coordinated and efficient manner.
A moving replication fork. (A) This schematic diagram shows a current view of the arrangement of replication proteins at a replication fork when the fork is moving. The diagram in Figure 5-21 has been altered by folding the DNA on the lagging strand to (more...)
On the lagging strand, the DNA replication machine leaves behind a series of unsealed Okazaki fragments, which still contain the RNA that primed their synthesis at their 5′ ends. This RNA is removed and the resulting gap is filled in by DNA repair enzymes that operate behind the replication fork (see ).
A Strand-directed Mismatch Repair System Removes Replication Errors That Escape from the Replication Machine
As stated previously, bacteria such as E. coli are capable of dividing once every 30 minutes, making it relatively easy to screen large populations to find a rare mutant cell that is altered in a specific process. One interesting class of mutants contains alterations in so-called mutator genes, which greatly increase the rate of spontaneous mutation when they are inactivated. Not surprisingly, one such mutant makes a defective form of the 3′-to-5′ proofreading exonuclease that is a part of the DNA polymerase enzyme (see and ). When this activity is defective, the DNA polymerase no longer proofreads effectively, and many replication errors that would otherwise have been removed accumulate in the DNA.
The study of other E. coli mutants exhibiting abnormally high mutation rates has uncovered another proofreading system that removes replication errors made by the polymerase that have been missed by the proofreading exonuclease. This strand-directed mismatch repair system detects the potential for distortion in the DNA helix that results from the misfit between noncomplementary base pairs. But if the proofreading system simply recognized a mismatch in newly replicated DNA and randomly corrected one of the two mismatched nucleotides, it would mistakingly “correct” the original template strand to match the error exactly half the time, thereby failing to lower the overall error rate. To be effective, such a proofreading system must be able to distinguish and remove the mismatched nucleotide only on the newly synthesized strand, where the replication error occurred.
The strand-distinction mechanism used by the mismatch proofreading system in E. coli depends on the methylation of selected A residues in the DNA. Methyl groups are added to all A residues in the sequence GATC, but not until some time after the A has been incorporated into a newly synthesized DNA chain. As a result, the only GATC sequences that have not yet been methylated are in the new strands just behind a replication fork. The recognition of these unmethylated GATCs allows the new DNA strands to be transiently distinguished from old ones, as required if their mismatches are to be selectively removed. The three-step process involves recognition of a mismatch, excision of the segment of DNA containing the mismatch from the newly synthesized strand, and resynthesis of the excised segment using the old strand as a template—thereby removing the mismatch. This strand-directed mismatch repair system reduces the number of errors made during DNA replication by an additional factor of 102 (see , p. 243).
A similar mismatch proofreading system functions in human cells. The importance of this system is indicated by the fact that individuals who inherit one defective copy of a mismatch repair gene (along with a functional gene on the other copy of the chromosome) have a marked predisposition for certain types of cancers. In a type of colon cancer called hereditary nonpolyposis colon cancer (HNPCC), spontaneous mutation of the remaining functional gene produces a clone of somatic cells that, because they are deficient in mismatch proofreading, accumulate mutations unusually rapidly. Most cancers arise from cells that have accumulated multiple mutations (discussed in Chapter 23), and cells deficient in mismatch proofreading therefore have a greatly enhanced chance of becoming cancerous. Fortunately, most of us inherit two good copies of each gene that encodes a mismatch proofreading protein; this protects us, because it is highly unlikely that both copies would mutate in the same cell.
In eucaryotes, the mechanism for distinguishing the newly synthesized strand from the parental template strand at the site of a mismatch does not depend on DNA methylation. Indeed, some eucaryotes—including yeasts and Drosophila—do not methylate any of their DNA. Newly synthesized DNA strands are known to be preferentially nicked, and biochemical experiments reveal that such nicks (also called single-strand breaks) provide the signal that directs the mismatch proofreading system to the appropriate strand in a eucaryotic cell ().
A model for strand-directed mismatch repair in eucaryotes. (A) The two proteins shown are present in both bacteria and eucaryotic cells: MutS binds specifically to a mismatched base pair, while MutL scans the nearby DNA for a nick. Once a nick is found, (more...)
DNA Topoisomerases Prevent DNA Tangling During Replication
As a replication fork moves along double-stranded DNA, it creates what has been called the “winding problem.” Every 10 base pairs replicated at the fork corresponds to one complete turn about the axis of the parental double helix. Therefore, for a replication fork to move, the entire chromosome ahead of the fork would normally have to rotate rapidly (). This would require large amounts of energy for long chromosomes, and an alternative strategy is used instead: a swivel is formed in the DNA helix by proteins known as DNA topoisomerases.
The “winding problem” that arises during DNA replication. For a bacterial replication fork moving at 500 nucleotides per second, the parental DNA helix ahead of the fork must rotate at 50 revolutions per second.
A DNA topoisomerase can be viewed as a reversible nuclease that adds itself covalently to a DNA backbone phosphate, thereby breaking a phosphodiester bond in a DNA strand. This reaction is reversible, and the phosphodiester bond re-forms as the protein leaves.
One type of topoisomerase, called topoisomerase I, produces a transient single-strand break (or nick); this break in the phosphodiester backbone allows the two sections of DNA helix on either side of the nick to rotate freely relative to each other, using the phosphodiester bond in the strand opposite the nick as a swivel point (). Any tension in the DNA helix will drive this rotation in the direction that relieves the tension. As a result, DNA replication can occur with the rotation of only a short length of helix—the part just ahead of the fork. The analogous winding problem that arises during DNA transcription (discussed in Chapter 6) is solved in a similar way. Because the covalent linkage that joins the DNA topoisomerase protein to a DNA phosphate retains the energy of the cleaved phosphodiester bond, resealing is rapid and does not require additional energy input. In this respect, the rejoining mechanism is different from that catalyzed by the enzyme DNA ligase, discussed previously (see ).
The reversible nicking reaction catalyzed by a eucaryotic DNA topoisomerase I enzyme. As indicated, these enzymes transiently form a single covalent bond with DNA; this allows free rotation of the DNA around the covalent backbone bonds linked to the (more...)
A second type of DNA topoisomerase, topoisomerase II, forms a covalent linkage to both strands of the DNA helix at the same time, making a transient double-strand break in the helix. These enzymes are activated by sites on chromosomes where two double helices cross over each other. Once a topoisomerase II molecule binds to such a crossing site, the protein uses ATP hydrolysis to perform the following set of reactions efficiently: (1) it breaks one double helix reversibly to create a DNA “gate;” (2) it causes the second, nearby double helix to pass through this break; and (3) it then reseals the break and dissociates from the DNA (). In this way, type II DNA topoisomerases can efficiently separate two interlocked DNA circles ().
A model for topoisomerase II action. As indicated, ATP binding to the two ATPase domains causes them to dimerize and drives the reactions shown. Because a single cycle of this reaction can occur in the presence of a non-hydrolyzable ATP analog, ATP hydrolysis (more...)
The DNA-helix-passing reaction catalyzed by DNA topoisomerase II. Identical reactions are used to untangle DNA inside the cell. Unlike type I topoisomerases, type II enzymes use ATP hydrolysis and some of the bacterial versions can introduce superhelical (more...)
The same reaction also prevents the severe DNA tangling problems that would otherwise arise during DNA replication. This role is nicely illustrated by mutant yeast cells that produce, in place of the normal topoisomerase II, a version that is inactive at 37°C. When the mutant cells are warmed to this temperature, their daughter chromosomes remain intertwined after DNA replication and are unable to separate. The enormous usefulness of topoisomerase II for untangling chromosomes can readily be appreciated by anyone who has struggled to remove a tangle from a fishing line without the aid of scissors.
DNA Replication Is Similar in Eucaryotes and Bacteria
Much of what we know about DNA replication was first derived from studies of purified bacterial and bacteriophage multienzyme systems capable of DNA replication in vitro. The development of these systems in the 1970s was greatly facilitated by the prior isolation of mutants in a variety of replication genes; these mutants were exploited to identify and purify the corresponding replication proteins. The first mammalian replication system that accurately replicated DNA in vitro was described in the mid-1980s, and mutations in genes encoding nearly all of the replication components have now been isolated and analyzed in the yeast Saccharomyces cerevisiae. As a result, a great deal is known about the detailed enzymology of DNA replication in eucaryotes, and it is clear that the fundamental features of DNA replication—including replication fork geometry and the use of a multiprotein replication machine—have been conserved during the long evolutionary process that separates bacteria and eucaryotes.
There are more protein components in eucaryotic replication machines than there are in the bacterial analogs, even though the basic functions are the same. Thus, for example, the eucaryotic single-strand binding (SSB) protein is formed from three subunits, whereas only a single subunit is found in bacteria. Similarly, the DNA primase is incorporated into a multisubunit enzyme called DNA polymerase α. The polymerase α begins each Okazaki fragment on the lagging strand with RNA and then extends the RNA primer with a short length of DNA, before passing the 3′ end of this primer to a second enzyme, DNA polymerase δ. This second DNA polymerase then synthesizes the remainder of each Okazaki fragment with the help of a clamp protein ().
A mammalian replication fork. The fork is drawn to emphasize its similarity to the bacterial replication fork depicted in Figure 5-21. Although both forks use the same basic components, the mammalian fork differs in at least two important respects. First, (more...)
As we see in the next section, the eucaryotic replication machinery has the added complication of having to replicate through nucleosomes, the repeating structural unit of chromosomes discussed in Chapter 4. Nucleosomes are spaced at intervals of about 200 nucleotide pairs along the DNA, which may explain why new Okazaki fragments are synthesized on the lagging strand at intervals of 100–200 nucleotides in eucaryotes, instead of 1000–2000 nucleotides as in bacteria. Nucleosomes may also act as barriers that slow down the movement of DNA polymerase molecules, which may be why eucaryotic replication forks move only one-tenth as fast as bacterial replication forks.
Summary
DNA replication takes place at a Y-shaped structure called a replication fork. A self-correcting DNA polymerase enzyme catalyzes nucleotide polymerization in a 5′-to-3′ direction, copying a DNA template strand with remarkable fidelity. Since the two strands of a DNA double helix are antiparallel, this 5′-to-3′ DNA synthesis can take place continuously on only one of the strands at a replication fork (the leading strand). On the lagging strand, short DNA fragments must be made by a “backstitching” process. Because the self-correcting DNA polymerase cannot start a new chain, these lagging-strand DNA fragments are primed by short RNA primer molecules that are subsequently erased and replaced with DNA.
DNA replication requires the cooperation of many proteins. These include (1) DNA polymerase and DNA primase to catalyze nucleoside triphosphate polymerization; (2) DNA helicases and single-strand DNA-binding (SSB) proteins to help in opening up the DNA helix so that it can be copied; (3) DNA ligase and an enzyme that degrades RNA primers to seal together the discontinuously synthesized lagging-strand DNA fragments; and (4) DNA topoisomerases to help to relieve helical winding and DNA tangling problems. Many of these proteins associate with each other at a replication fork to form a highly efficient “replication machine,” through which the activities and spatial movements of the individual components are coordinated.