

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Panayiotopoulos CP. The Epilepsies: Seizures, Syndromes and Management. Oxfordshire (UK): Bladon Medical Publishing; 2005.

Neonatal Seizures
Clinical noteNeonatal seizures or neonatal convulsions are epileptic fits occurring from birth to the end of the neonatal period. 1–18 The neonatal period is the most vulnerable of all periods of life for developing seizures, particularly in the first 1–2 days to the first week from birth. They may be short-lived events lasting for a few days only. However, they often signify serious malfunction of or damage to the immature brain and constitute a neurological emergency demanding urgent diagnosis and management.
Demographic Data
The prevalence is approximately 1.5% and overall incidence approximately 3 per 1000 live births. The incidence in pre-term infants is very high (57–132 per 1000 live births). Most (80%) neonatal seizures occur in the first 1–2 days to the first week of life.
Clinical Manifestations
Neonatal seizures, as with any other type of seizure, are paroxysmal, repetitive and stereotypical events. They are usually clinically subtle, inconspicuous and difficult to recognise from the normal behaviours of the inter-ictal periods or physiological phenomena. There is no recognisable post-ictal state. Generalised tonic clonic seizures (GTCS) are exceptional. The most widely used scheme is by Volpe20 of five main types of neonatal seizure.
- Subtle seizures (50%)
- Tonic seizures (5%)
- Clonic seizures (25%)
- Myoclonic seizures (20%)
- Non-paroxysmal repetitive behaviours
Useful Definitions
The neonatal period is defined as the first 28 days of life of a full-term infant.
Neonatal seizures are those that occur from birth to the end of the neonatal period.
Gestational age is defined as the duration of pregnancy.
Chronological age is the actual legal age of the infant from the time of birth.
Conceptional age is the combined gestational and chronological ages.
Full-term infants are those of 40 weeks gestational age.
ILAE Classification and Definitions
The ILAE Commission (1989)19 broadly classifies neonatal seizures amongst ‘epilepsies and syndromes undetermined as to whether they are focal or generalised’ under the subheading ‘with both generalised and focal seizures’.
Neonatal seizures differ from those of older children and adults. The most frequent neonatal seizures are described as subtle because the clinical manifestations are frequently overlooked. These include tonic, horizontal deviation of the eyes with or without jerking, eyelid blinking or fluttering, sucking, smacking or other oral–buccal–lingual movements, swimming or pedalling movements and, occasionally, apnoeic spells. Other neonatal seizures occur as tonic extension of the limbs, mimicking decerebrate or decorticate posturing. These occur particularly in premature infants. Multifocal clonic seizures characterised by clonic movements of a limb, which may migrate to other body parts or other limbs or focal clonic seizures, which are much more localised, may occur. In the latter, the infant is usually not unconscious. Rarely, myoclonic seizures may occur and the EEG pattern is frequently that of suppression–burst activity. The tonic seizures have a poor prognosis because they frequently accompany intraventricular haemorrhage. The myoclonic seizures also have a poor prognosis because they are frequently a part of the early myoclonic encephalopathy syndrome.19
In another scheme by Mizrahi12 neonatal seizures are classified as follows: focal clonic, focal tonic, generalised tonic, myoclonic, spasms and motor automatisms (which include occular signs, oral–buccal–lingual movements, progression movements and complex purposeless movements).
Nearly one-quarter of infants experience several seizure types and the same seizure may manifest with subtle, clonic, myoclonic, autonomic or other symptoms (Figure 5.1).

Figure 5.1
Ictal EEG patterns in a 2-day-old boy with right middle cerebral artery thrombosis Top and middle: Apnoeic, myoclonic, clonic and subtle seizure of motor automatisms associated with various ictal EEG patterns and locations.
Subtle Seizures
Subtle seizures are far more common than other types of neonatal seizures. They are described as subtle because the clinical manifestations are frequently overlooked. They imitate normal behaviours and reactions. These include the following.
- Ocular movements, which range from random and roving eye movements to sustained conjugate tonic deviation with or without jerking. Eyelid blinking or fluttering, eyes rolling up, eye opening, fixation of a gaze or nystagmus may occur alone or with other ictal manifestations.
- Oral–buccal–lingual movements (sucking, smacking, chewing and tongue protrusions).
- Progression movements (rowing, swimming, pedalling, bicycling, thrashing or struggling movements).
- Complex purposeless movements (sudden arousal with episodic limb hyperactivity and crying).15
Motor Seizures
Clonic seizures are rhythmic jerks that may localise in a small part of the face or limbs, axial muscles and the diaphragm or be multifocal or hemiconvulsive. Todd’s paresis follows prolonged hemiconvulsions.
Tonic seizures manifest with sustained contraction of facial, limb, axial and other muscles. They may be focal, multifocal or generalised, symmetrical or asymmetrical. Truncal or limb tonic extension imitates decerebrate or decorticate posturing.
Myoclonic seizures are rapid, single or arrhythmic repetitive jerks. They may affect a finger, a limb or the whole body. They may mimic the Moro reflex and startling responses. They are more frequent in pre-term than full-term infants indicating, if massive, major brain injury and poor prognosis. Myoclonic seizures are associated with the most severe brain damage.21 However, healthy pre-term and rarely full-term neonates may have abundant myoclonic movements during sleep. Neonates have cortical, reticular and segmental types of myoclonus, similar to adult forms.22
Spasms producing flexion or extension similar to those of West syndrome are rare. They are slower than myoclonic seizures and faster than tonic seizures.
Autonomic Ictal Manifestations
Autonomic ictal manifestations commonly occur with motor manifestations in 37% of subtle seizures.23,24 These are paroxysmal changes of heart rate, respiration and systemic blood pressure.25,26 Apnoea as an isolated seizure phenomenon unaccompanied by other clinical epileptic features is probably exceptional.26 Salivation and pupillary changes are common.
Duration of Neonatal Seizures
The duration of neonatal seizures is usually brief (10 s to 1–2 min) and repetitive with a median of 8 min in between each seizure. Longer seizures and status epilepticus develop more readily at this age, but convulsive neonatal status epilepticus is not as severe as that of older infants and children.
Non-Epileptic Neonatal Seizures
By definition all neonatal seizures are epileptic in origin, generated by abnormal, paroxysmal and hypersynchronous neuronal discharges characteristic of epileptogenesis.
The characterisation of neonatal seizures as epileptic and non-epileptic by Kellaway and Mizrahi4,10,27 is a topic of considerable debate.
Epileptic Neonatal Seizures
Focal clonic, focal tonic and some types of myoclonic jerks are genuine epileptic seizures documented with ictal EEG changes and they have a high correlation with focal brain lesions and a favourable short-term outcome.4,10,27
Non-Epileptic Neonatal Seizures
Many of the subtle seizures, generalised tonic posturing and some myoclonic symptoms may be non-epileptic seizures. These show clinical similarities to reflex behaviours of the neonates, they are not associated with ictal EEG discharges and are commonly correlated with diffuse abnormal brain processes such as hypoxic–ischaemic encephalopathy and a poor short-term outcome.4,10,27 They are considered as exaggerated reflex behaviours due to abnormal release of brain stem tonic mechanisms from cortical control. Hence, the term brain stem release phenomena:4,27 “They most typically occur in neonates with clinical and EEG evidence of forebrain depression that may release brain stem facilitatory centres for generating reflex behaviours without cortical inhibition”.4,10,27
The argument to support the non-epileptic nature of these episodic clinical events is that they have the following characteristics.4,10,27
- They are suppressed by restraint or repositioning of the infant.
- They are elicited by tactile stimulation and their intensity is proportional to the rate, intensity and number of sites of stimulation. Stimulation at one site can provoke paroxysmal movements at another site.
- They are not associated with ictal EEG discharges.
Reservations about these features are based on the following reasons.
- The electrical seizure activity may occur deep within brain structures that are inaccessible to an EEG. This is well documented in neurosurgical patients with simultaneous surface and deep EEG recordings.
- The responses to stimulation and restraint are also well-known phenomena of genuine epileptic seizures (see photic and tactile stimulation and inhibition).
Aetiology
Aetiology of neonatal seizures is extensive and diverse (Table 5.1). Severe causes predominate. The prevalence and significance of aetiological factors are continuously changing and differ between developed and developing countries depending on available improved neonatal and obstetric care.
Table 5.1
Main causes of neonatal seizures
By far the commonest cause is hypoxic–ischaemic encephalopathy. It may be responsible for 80% of all seizures in the first 2 days of life.28 Brain damage due to prenatal distress and malformations of cortical development is being increasingly recognised. Intracranial haemorrhage and infarction, stroke29 and prenatal and neonatal infections are common. Most previously common acute metabolic disturbances such as electrolyte and glucose abnormalities have been minimised because of improved neonatal intensive care and awareness of nutritional hazards. Late hypocalcaemia is virtually eliminated, while electrolytic derangement and hypoglycaemia are now rare.
Inborn errors of metabolism such as urea cycle disorders are rare.10,20,30,31
Pyridoxine dependency, with seizures in the first days of life (which are reversible with treatment), is exceptional.
Exogenous causes of neonatal convulsions may be iatrogenic or due to drug withdrawal in babies born to mothers on drugs.24
In most cases, the neonate may present with a combination of different neurological disturbances, each of which can cause seizures.
Pathophysiology
The early postnatal development time is a period of increased susceptibility to seizures in relation to other ages. This may be due to a combination of factors specific to the developing brain that enhance excitation and diminish inhibition. There is an unequal distribution of anticonvulsant and proconvulsant neuro-transmitters and networks.13,32,33
Animal studies are contradictory regarding the effect of prolonged epileptic seizures on the developing immature brain.34–36
See also the pathophysiology of individual neonatal syndromes detailed in this section.
Diagnostic Procedures
Neonatal seizures represent one of the very few emergencies in the newborn. Abnormal, repetitive and stereotypic behaviours of neonates should be suspected and evaluated as possible seizures. Polygraphic video–EEG recording of suspected events is probably mandatory for an incontrovertible seizure diagnosis. Confirmation of neonatal seizures should initiate urgent and appropriate clinical and laboratory evaluation for the aetiological cause (Table 5.1) and treatment. Family and prenatal history is important. A thorough physical examination of the neonate should be coupled with urgent and comprehensive biochemical tests for correctable metabolic disturbances. Although rare, more severe inborn errors of metabolism should be considered for diagnosis and treatment.
Brain Imaging
Cranial ultrasonography, brain imaging with X-ray computed tomography (CT) scan and preferably magnetic resonance imaging (MRI)37 should be used for the detection of structural abnormalities such as malformations of cortical development, intracranial haemorrhage, hydrocephalus and cerebral infarction.
Cranial ultrasonograghy is the main imaging modality of premature neonates and well suited for the study of neonates in general. It is performed at the bedside and provides effective assessment of ventricular size and other fluid-containing lesions as well as effective viewing of haemorrhagic and ischaemic lesions and their evolution. Cranial ultrasonography is limited in resolution and the type of lesions that it can identify.
A CT brain scan is often of secondary or adjunctive importance to ultrasound. Last-generation CT brain scan images are of high resolution, can be generated within seconds and can accurately detect haemorrhage, infarction, gross malformations and ventricular and other pathological conditions. A CT scan has low sensitivity in many other brain conditions such as abnormalities of cortical development where MRI is much superior. However, MRI interpretation should take into consideration the normal developmental and maturational states of neonates and infants. In infants younger than 6 months, cortical abnormalities are detected with T2-weighted images, whereas T1-weighted images are needed for the evaluation of brain maturation.37
Electroencephalography
The neonatal EEG is probably one of the best and most useful of EEG applications.10,38–45 However, neonatal EEG recordings and interpretations require the special skills of well-trained technologists and physicians. Polygraphic studies with simultaneous video–EEG recording are essential.46
Only 10% of neonates with suspected seizures have EEG confirmation. Suspected clonic movements have the highest yield of 44% but this is only 17% for ‘subtle’ movements.47
Inter-Ictal EEG
Inter-ictal EEG epileptogenic spikes or sharp slow wave foci are not reliable markers at this age.
Certain inter-ictal EEG patterns may have diagnostic significance (Figure 5.2 and 5.3.)4,10
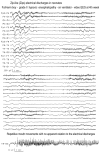
Figure 5.2
Zip-like electrical discharges Top and middle: Continuous recording in a neonate with severe brain hypoxia. Zip-like electrical discharges consist of high frequency rapid spikes of accelerating and (more...)
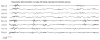
Figure 5.3
Inter-ictal EEG in a neonate with benign neonatal (non-familial) seizures. The theta pointu alternant pattern is usually associated with a good prognosis.
- Electrocerebral inactivity of a flat or near-flat EEG of severe brain damage.
- The suppression–burst pattern of neonatal epileptic encephalopathies (Figure 5.4).
- Theta pointu alternant of benign neonatal convulsions (Figure 5.3).
- Persistently focal sharp or slow waves in localised lesions.
- Quasi-periodic focal or multifocal pattern in neonatal herpes simplex encephalitis.48
- Inter-hemispheric or intra-hemispheric abnormalities.

Figure 5.4
The burst–suppression pattern in a neonate with severe hypoxic encephalopathy.
Background EEG activity, mainly in serial EEGs, often provides objective evidence of the degree and severity of the underlying cause.4,10,31,49
Important Note on the Suppression–burst Pattern
The suppression–burst pattern is relatively frequent in the neonatal period. It is associated with heterogeneous seizures and can be induced by drugs.50,51 It is common in neonatal ischaemic encephalopathy where it is usually transient and short lived.50 Conversely, the suppression–burst pattern is relatively stable lasting for more than 2 weeks in Ohtahara syndrome and early myoclonic encephalopathy.52
Ictal EEG
Documentation of seizures with an ictal EEG is often mandatory in view of the subtle clinical seizure manifestations (Figures 5.1 and 5.2). EEG ictal activity may be focal or multifocal appearing in a normal or abnormal background. The electrical ictal seizure EEG patterns of neonatal seizures vary significantly even in the same neonate and in the same EEG recording (Figure 5.1). The same EEG may show focal or multifocal ictal discharges that may occur simultaneously or independently in different brain locations.
Ictal EEG paroxysms consist of repetitive waves with a predominant beta, alpha, theta and delta range or a mixture of all that may accelerate in speed, decelerate in speed or both (Figures 5.1 and 5.2). These are spikes, sharp or saw tooth or sinusoidal waves (monomorphic or polymorphic) ranging in amplitude from very low to very high. The patterns may be synchronous or asynchronous, focal or multifocal and, less frequently, generalised. They may appear and disappear suddenly or build up from accelerating localised repetitive waves. Ictal discharges may gradually or abruptly change in frequency, amplitude and morphology in the course of the same or subsequent seizures. Conversely, they may remain virtually unchanged from onset to termination. The background EEG may be normal or abnormal.
Definition of Zips
Zips. This is a descriptive term I coined for a common ictal EEG pattern in neonates which consists of localised episodic rapid spikes of accelerating and decelerating speed that look like zips (Figure 5.2). Zips may be associated with subtle and focal clonic/tonic seizures or remain clinically silent. Zips of subtle seizures are often multifocal and of shifting localisation.
Focal EEG ictal discharges are usually associated with subtle, clonic or tonic seizures. The most common locations are the centrotemporal followed by the occipital regions. Midline (Cz) and temporal regions may be involved but frontal localisations are exceptional. The same infant may have unifocal or multifocal ictal discharges that may be simultaneous, develop one from the other or occur independently in different brain sites. Neonates do not show the clinical or EEG Jacksonian march of older children. There may be abrupt changes of location in the progress of the seizure.
Seizures with consistently focal EEG paroxysms are highly correlated with focal brain lesions. Seizures that lack or have an inconstant relationship with EEG discharges correlate with diffuse pathological conditions.
Generalised manifestations are more likely to occur with myoclonic jerks and neonatal spasms.
Two specific electrical seizure patterns of neonates usually carry a poor prognosis because they are ‘typically associated with severe encephalopathies’.10,27,53,54
- Alpha seizure discharges are characterised by sustained and rhythmic activity of 12 Hz and 20–70 μV in the centrotemporal regions.
- Electrical seizure activity of the depressed brain is of low voltage and long duration. It is highly localised on one side and shows little tendency to spread.
Post-Ictal EEG
Post-ictally, the EEG usually returns to the pre-ictal state immediately (Figure 5.1). Transient slowing or depression of EEG activity may occur following frequent or prolonged seizures.
Stimulus-Evoked Electrographic Patterns
Stimulus-evoked electrographic patterns with or without concomitant clinical ictal manifestations usually occur in pre-term neonates or neonates with significant diffuse or multifocal brain damage.55 The electrographic seizures are elicited by tactile or painful stimulation. The majority of neonates die or have significant neurological handicaps.
Electroclinical Dissociation or Decoupling Response
Only one-fifth (21%) of electrical ictal EEG patterns (electrical or electrographic seizures) associate with distinctive clinical manifestations (electroclinical seizures). All others are occult, that is they are clinically silent or subclinical.56
Electrographic or electrical seizures, namely EEG electrical seizure activity without apparent clinical manifestations, are more common after initiation of anti-epileptic drugs (AEDs). This is because AEDs may suppress the clinical manifestations of seizures but not the EEG ictal discharge. This phenomenon is named the ‘decoupling response’ 10,27,53 or ‘electroclinical dissociation’.57,58 Electroclinical dissociation may arise from foci not consistently reflected in surface electrodes.57
Neonates with electrographic seizures do not differ from those with exclusively electroclinical seizures regarding peri-natal factors, aetiology or outcome, though the background EEG is more abnormal in the electrographic group.57 Movements of the limbs occur at a statistically significant higher rate during electroclinical seizures. Electrographic seizures, like electroclinical seizures, are also associated with disturbed cerebral metabolism.59
Prognosis
This is cause dependent because the main factor that determines outcome is the underlying cause and not the seizures themselves. Despite high mortality (approximately 15%) and morbidity (approximately 30%), one-half of neonates with seizures achieve a normal or near normal state. One-third of the survivors develop epilepsy.60 Table 5.2 provides indicators of good, bad or intermediate prognosis.
Table 5.2
Indicators of prognosis
Differential Diagnosis
Neonatal seizures often impose significant difficulties in their recognition and differentiation from normal or abnormal behaviours of the pre-term and full-term neonate.4,10
Important noteAs a rule any suspicious repetitive and stereotypical events should be considered as possible seizures requiring video–EEG recording confirmation.
Normal Behaviours
Amongst normal behaviours neonates may stretch, exhibit spontaneous sucking movements and have random and non-specific movements of the limbs. Intense physiological myoclonus may occur during rapid eye movement sleep. Jitteriness or tremulousness of the extremities or facial muscles is frequent in normal or abnormal neonates.
Tremor has a symmetrical ‘to and fro’ motion, is faster than clonic seizures, mainly affects all four limbs and will stop when the limb is restrained or repositioned. Conversely, clonic seizures are mainly focal, usually have a rate of 3–4 Hz or slower, decelerate in the progress of the attack and they are not interrupted by passive movements.
Abnormal Behaviours
Amongst abnormal behaviours of neonates with CNS disorders are episodic and repetitive oral–buccal–lingual movements. These are often reproducible with tactile or other stimuli and are interrupted by restraint. Conversely, neonatal seizures persist despite restraint and they are rarely stimulus sensitive.
Non-Epileptic Movement Disorders
Neonatal seizures should be differentiated from benign neonatal sleep myoclonus, hyperekplexia and other non-epileptic movement disorders.
Significant impairment of vital signs, which may be periodic, is mainly due to non-neurological causes. Changes in respiration, heart rate and blood pressure are exceptional sole manifestations of neonatal seizures.
Inborn errors of metabolism manifest with neonatal subtle seizures or abnormal movements that may not be genuine epileptic seizures. Their identity is often revealed by other associated significant symptoms, such as peculiar odours, protein intolerance, acidosis, alkalosis, lethargy or stupor. In most cases, pregnancy, labour and delivery are normal. Food intolerance may be the earliest indication of a systemic abnormality. If untreated, metabolic disorders commonly lead to lethargy, coma and death. In surviving infants weight loss, poor growth and failure to thrive are common.
Management 14,17,61
This demands accurate aetiological diagnosis and treatment of the cause of the seizures. The principles of general medical management and cardiovascular and respiratory stabilisation should be early and appropriately applied. Cardiorespiratory symptoms may result from the underlying disease, the seizures and the anti-epileptic medication.
Neonatal seizures of metabolic disturbances need correction of the underlying cause and not anti-epileptic medication. A trial of pyridoxine may be justifiable.
The drug treatment of neonatal seizures is empirical with significant practice variations amongst physicians. Phenobarbitone first and then phenytoin are the most commonly used AEDs, although short-acting benzodiazepines are gaining ground. Large loading doses are followed by a maintenance scheme for a variable period.
Clinical noteFacts and requirements for the treatment of neonatal seizures
Neonatal seizures have a high prevalence and their response to anti-epileptic drugs (AEDs) is likely to be different to that of other specified groups of patients. Yet, current treatment of neonatal seizures is entirely empirical. Neonatologists rely on their medical judgment and “trials by success and error” with off-label use of new and old AEDs.
The authorities, including formal regulatory agents, should urgently address these issues.
Phenobarbitone and Phenytoin
Phenobarbitone and phenytoin are equally but incompletely effective as anticonvulsants in neonates. Phenobarbitone in a loading dose of 15–20 mg/kg and a maintenance dose of 3–4 mg/kg daily controls one-third of neonatal seizures.62 Efficacy may improve to 85% with stepwise increments to 40 mg/kg.63 The serum levels required are between 16 and 40 μg/ml. Phenytoin may be equally as effective as phenobarbitone at a loading dose of 15–20 mg/kg.64 With either drug given alone, the seizures are controlled in fewer than half of the neonates.64 The severity of the seizures appears to be a stronger predictor of the success of treatment than the assigned AED. Mild seizures or seizures decreasing in severity before treatment are more likely to respond regardless of the treatment assignment.
Fosphenytoin
Fosphenytoin is an attractive alternative to phenytoin because of less potential for adverse reactions at the infusion site and the facility for intramuscular administration.15 This needs further evaluation.
Other Drugs
Intravenous benzodiazepines such as diazepam, lorazepam,65 clonazepam and midazolam66 are used particularly in Europe for acute neonatal seizures. They may be used as the first anti-seizure AED. However, in a recent randomised trial of second-line anticonvulsant treatments for neonates,67 11 out of 22 subjects responded to phenobarbitone at a dose of 40 mg/kg as first-line treatment. Three of five neonates treated with lignocaine responded. However, of 6 neonates treated with benzodiazepines as second-line treatment, none responded and their neurodevelopmental outcome was poor.67
Primidone, valproate, lignocaine,68 carbamazepine 69 and paraldehyde (now not available in the USA) are also used mainly as adjunctive AEDs if others fail. Of the new AEDs vigabatrin 70 and lamotrigine 71 may be effective.
Maintenance Treatment
Maintenance treatment may not be needed or be brief as the active seizure period in neonates is usually short. Less than 15% of infants with neonatal seizures will have recurrent seizures after the newborn period.72 A normal EEG73 and other predictors of good outcome74 may encourage early discontinuation of treatment. The current trend is to withdraw the AED 2 weeks after the last seizure.
Clinical noteDo electrographic (electrical) seizures need treatment?
There is significant difference of opinion as to whether EEG electrical seizure activity that may persist despite drug control of clinical seizures needs more vigorous treatment. Electrical seizures may be highly resistant to drug treatment and attempts to eliminate them may require high doses of usually multiple drugs with significant adverse reactions such as CNS or respiratory depression and systemic hypotension. The risks should be weighed against the benefits while also remembering that these will eventual subside in time.
Neonatal Syndromes
Despite the high prevalence of neonatal seizures, epileptic syndromes in neonates are rare. Four syndromes have been recognised by the ILAE.19,75
- Benign familial neonatal seizures
- Benign neonatal seizures (non-familial)
- Early myoclonic encephalopathy
- Ohtahara syndrome
Benign Familial Neonatal Seizures
Benign familial neonatal seizures 76–80 constitute a rare autosomal dominant epileptic syndrome characterised by frequent brief seizures within the first days of life.
Demographic Data
Onset is commonly in the first week of life, mainly on the second or third day.81 All 116 affected individuals in one study had seizures by 2–8 days of life.81 The strict age dependence of the syndrome is indicated by the fact that affected premature babies develop seizures later.6 Contrary to this, in one-third of patients seizures may start as late as 3 months of age.82 Boys and girls are equally affected. The syndrome appears to be rare but this may be under-recognised or not reported by the families who know its benign character from their own experience. So far 44 families with 355 affected members have been reported.76 The incidence is 14.4 per 100 000 live births.6
Changes in the New ILAE Diagnostic Scheme regarding the Nomenclature and Classification of Neonatal Syndromes
The 1989 ILAE classification considers ‘benign familial neonatal convulsions’ and ‘benign neonatal convulsions (non-familial)’ as ‘idiopathic generalised epilepsies (age related)’.19
The ILAE new diagnostic scheme abandons the name convulsions using instead seizures, thus also emphasising that these are ‘conditions with epileptic seizures that do not require a diagnosis of epilepsy’.75
‘Early myoclonic encephalopathy’ and ‘Ohtahara syndrome’ are considered as ‘generalised symptomatic epilepsies of non-specific aetiology (age related)’19 by the 1989 ILAE classification. The new ILAE diagnostic scheme classified them as ‘epileptic encephalopathies (in which the epileptiform abnormalities may contribute to progressive dysfunction)’
ILAE Definition for Benign Familial Neonatal Convulsions
Benign familial neonatal convulsions are a rare, dominantly inherited disorder manifesting mostly on the second and third days of life, with clonic or apnoeic seizures and no specific EEG criteria. History and investigations reveal no aetiological factors. Approximately 14% of these patients later develop epilepsy.19
Clinical Manifestations
Seizures mainly occur in full-term normal neonates after a normal pregnancy and delivery and without precipitating factors. Seizures are brief, of 1–2 min and may be as frequent as 20–30 per day.
Most seizures start with tonic motor activity and posturing with apnoea followed by vocalisations, ocular symptoms, other autonomic features, motor automatisms, chewing and focal or generalised clonic movements.76,77,83,84 The clonic components of the later phase are usually asymmetrical and unilateral. The post-ictal state is brief and inter-ictally the neonates are normal.
Pure clonic or focal seizures are considered rare.
Aetiology
This is a genetically determined channelopathy of an autosomal dominant pattern of inheritance and a high degree (approximately 85%) of penetrance. Thus, 15% of those with a mutant gene are not clinically affected. The disease is caused by mutations in the voltage-gated potassium channel subunit gene KCNQ2 on chromosome 20q 13.384,85 and KCNQ3 on chromosome 8q.13.3.86–91 More than 11 mutations have been identified in KCNQ2, but only two have been identified in KCNQ3.92,93 Mutations in either KCNQ2 or KCNQ3 can produce the same phenotype.
Mutations in the sodium channel subunit gene SCN2A appear specific to ‘benign familial neonatal-infantile seizures’.* 94
Pathophysiology
It appears that benign familial neonatal seizures are caused by a small loss of function of heteromeric voltage-gated potassium channels that decrease the potassium current. This impairs repolarisation of the neuronal cell membrane resulting in hyperexcitability of the brain that can produce seizures. It has also been postulated that a slight reduction in KCNQ channels alone cannot produce seizure activity, but can facilitate it under conditions of unbalanced neurotransmission, either by an increase in excitation or decrease in inhibition.96 Thus, this unbalance in excitation and inhibition could be one possible explanation as to why the neonatal period is a vulnerable time for the seizures to occur. Another possibility is the differential expression of potassium channels during different stages of maturation.80
Diagnostic Procedures
All relevant biochemical, haematological and metabolic screenings and brain imaging are normal.
Electroencephalography
The inter-ictal EEG may be normal, discontinuous, have focal or multifocal abnormalities or have a theta pointu alternant pattern (Figure 5.3). The inter-ictal EEG is of limited value though it may exclude other causes of serious neonatal seizures.
The ictal EEG commonly starts with a synchronous and bilateral flattening of 5–19 s coinciding with apnoea and tonic motor activity. This is followed by bilateral and often asymmetrical discharges of spikes and sharp waves with a duration of 1–2 min, which coincide with vocalisations, chewing and focal or generalised clonic activity.76,77
Conversely, seizure manifestations and the ictal EEG indicated focal seizures in a neonate,97 had both focal and generalised features in another neonate98 and had right frontal onset with or without generalisation in a third neonate.99
Differential Diagnosis
A family history of similar convulsions, a prerequisite for the diagnosis of benign familial neonatal seizures, eliminates the possibility of other diseases. However, other causes of neonatal seizures should be excluded.
Despite artificially similar names3,75 benign familial neonatal seizures are entirely different from benign neonatal seizures (non-familial) (Table 5.3).
Table 5.3
Benign (non-familial) neonatal seizures versus benign familial neonatal seizures
Prognosis
Seizures remit between 1 and 6 months from onset; in 68% during the first 6 weeks. However, 10–14% may later develop other types of febrile (5%) or afebrile seizures. Afebrile seizures are not well defined in the relevant reports but they are probably heterogeneous. Idiopathic generalised seizures are more common. There have also been accounts of Rolandic seizures.
In some families none of the patients had seizures after the first 10 months of life, with long-term follow-up ranging from 10 months to 56 years.100 In other reports a few patients101 or 20%102 continued having seizures in adult life. The subsequent risk of a recurrent seizure disorder depends on whether other affected relatives developed a seizure disorder later in life.102
The prevalence of mental retardation and learning disability is reported to be approximately 2.5%, which is not significantly different from the expected rate for the general population.81
Deaths during neonatal seizures are exceptional but they have been reported.101
Management
There is no consensus regarding treatment. Convulsions usually remit spontaneously without medication. The use of anti-epileptic medication does not influence the eventual outcome. Prolonged seizures may be shortened or terminated with benzodiazepines, phenobarbitone or phenytoin.
The family’s reaction to and their fears about this inherited disease as well as means of appropriate consultations in order to reduce their magnitude should be appropriately considered.103
Patient noteA normal boy started having frequent seizures at age 3 days. He had 4–8 stereotyped fits per 24 hours, awake or asleep. All seizures started with tonic motor activity and posturing with apnoea for 5–10 s followed by vocalisations, ocular symptoms, other autonomic features, motor automatisms, chewing and focal or generalised, asymmetrical, occasionally unilateral clonic movements.
All relevant tests including an inter-ictal EEG were normal.
Recommended treatment with valproate was vigorously rejected by the grandmother, a dominant member of the family, who herself, her father and two of her 4 children had similar neonatal seizures without any residuals or consequences in their successful lives. The boy continued having his habitual seizures up to the age of 6 weeks. He was normal in between the fits. On last follow-up at age 2 years he is an entirely normal child for his age. “The granny was right again” the family admitted.
Benign Neonatal Seizures (Non-Familial)
Benign neonatal seizures (non-familial)76,79,104–106 constitute a short-lived and self-limited benign epileptic syndrome. It manifests with a single episode of repetitive lengthy seizures, which constitutes clonic status epilepticus.
Demographic Data
The age at onset is characteristically between the first and seventh days of life. It is by far more common (90%) between the fourth and sixth days, for which the synonym ‘fifth day fits’ was coined.104 Boys (62%) are slightly more affected than girls. The prevalence is 7% of neonatal seizures, but this has declined significantly in recent years.76,105
Clinical Manifestations
There is a one-off event of a repetitive lengthy seizure which constitutes clonic status epilepticus, which occurs in otherwise normal full-term neonates. This consists of successive unilateral clonic convulsions affecting the face and the limbs. Convulsions may change side and may also less often be bilateral. Apnoea is a common concomitant in one-third of these clonic fits. Each seizure lasts for 1–3 min repeating at frequent intervals and cumulating to discontinuous or continuous clonic status epilepticus. The whole seizure–status event lasts for 2 h to 3 days with a median of approximately 20 h. It does not recur again. Tonic seizures are incompatible with this syndrome.
Aetiology
This is unknown but is probably environmental. There is no genetic background and there are various other propositions:
- Environmental causes because of significant periodic variations in the prevalence of the syndrome.
- Acute zinc deficiency detected in the cerebrospinal fluid of affected neonates.
- Viral illness, mainly rotavirus.
- Type of feeding.
ILAE Definition and Corrections
Benign neonatal seizures (non-familial) are defined as follows in the 1989 ILAE classification.19
Benign neonatal convulsions are very frequently repeated clonic or apnoeic seizures occurring at about the fifth day of life, without known aetiology or concomitant metabolic disturbance. The inter-ictal EEG often shows alternating sharp theta waves. There is no recurrence of seizures and the psychomotor development is not affected.19
Comment. In the 1989 ILAE classification, benign neonatal seizures (non-familial) were classified as idiopathic generalised epilepsy (IGE) (age related).19 However, many authors have emphasised that this is a predominantly focal (and not generalised) seizure syndrome8,107 and this should be considered in future revisions. This syndrome most likely belongs to the category of “conditions with epileptic seizures that do not require a diagnosis of epilepsy”.75
Pathophysiology
The pathophysiology is unknown. However, this syndrome provides firm evidence that even prolonged seizures in early life may not produce hippocampal damage in the absence of other complicating factors.108
Diagnostic Procedures
By definition all tests other than EEG are normal.
Electroencephalography
The inter-ictal EEG shows a ‘theta pointu alternant pattern’ in one-half of cases (Figure 5.3).76,79 In the others the EEG may show focal or multifocal, non-specific abnormalities or a discontinuous pattern or it may be normal in approximately 10%.
The theta pointu alternant pattern consists of runs of a dominant theta wave activity of 4–7 Hz intermixed with sharp waves often of alternating side.76 It is not reactive to various stimuli. It may occur on awakening and during sleep. It may persist for 12 days after the cessation of convulsions. However, the theta pointu alternant pattern is not specific as it may be recorded in other conditions such as hypocalcaemia, meningitis, subarachnoid haemorrhage, in neonatal encephalopathies including hypoxic–ischaemic encephalopathy and benign familial neonatal seizures.
In follow-up studies, centrotemporal spikes are found at a later age in otherwise asymptomatic cases.
The ictal EEG consists of rhythmic spikes or slow waves mainly in the Rolandic regions though they can also localise anywhere else.76,105,107 The EEG ictal paroxysms may be unilateral, generalised or first localised and then generalised. The duration of the ictal discharges is 1–3 min and this may be followed by subclinical discharges for many hours.
Differential Diagnosis
The diagnosis can be made only after other causes of neonatal seizures have been excluded. The aetiologies of neonatal seizures with favourable outcomes include late hypocalcaemia, subarachnoid haemorrhage and certain meningitides (Table 5.2).76,105,107 There are significant differences between benign neonatal seizures (non-familial) and benign familial neonatal seizures despite the artificially similar names3,75 and the fact that they have a similar age at onset (Table 5.3).
Clinical noteDiagnostic tips for benign neonatal seizures (non-familial)
A single episode of repetitive clonic seizures that are mainly unilateral, often of alternating side and lasting for around 20 h in a full-term neonate that was normal up to that stage. All relevant investigations other than EEG are normal.
Prognosis
The prognosis is commonly excellent with normal development and no recurrence of seizures. Minor psychomotor deficits and occasional febrile or afebrile seizures (0.5%) have been reported.76,79,107 However, North et al.109 had less optimistic results. Afebrile seizures or developmental delay occurred in one-half of their patients and there was a single case of sudden infant death.109
Management
Convulsions usually remit spontaneously without medication. Prolonged seizures may be shortened or terminated by intravenous administration of benzodiazepines, phenobarbitone or phenytoin. If medications are used they are discontinued soon after the seizures subside.
Early Myoclonic Encephalopathy
Early myoclonic encephalopathy is a dreadful but fortunately rare epileptic encephalopathy of the first days and weeks of life. 50,52,110–113
Demographic Data
Early myoclonic encephalopathy usually starts in the first days of life, sometimes immediately after birth. More than 60% start before 10 days of age and rarely after the second month. Boys and girls are equally affected. The prevalence and incidence is unknown. There are approximately 80 reported cases, but this may be an underestimation because neonates with such a severe disease and early death may escape clinico-EEG diagnosis.
Clinical Manifestations
The syndrome manifests with a triad of intractable seizures. Erratic myoclonus appears first followed by simple focal seizures and later by tonic epileptic (infantile) spasms.
ILAE Definitions
Early myoclonic encephalopathy is defined as follows by the ILAE Commission (1989).19
The principal features of early myoclonic encephalopathy are onset occurring before age 3 months, initially fragmentary myoclonus and then erratic focal seizures, massive myoclonias or tonic spasms. The EEG is characterised by suppression–burst activity, which may evolve into hypsarrhythmia. The course is severe, psychomotor development is arrested and death may occur in the first year. Familial cases are frequent and suggest the influence of one or several congenital metabolic errors, but there is no specific genetic pattern.19
Clarifications on Myoclonus
There is no generally accepted, precise definition of “myoclonus” and there is a long-standing source of confusion and debate regarding the term and concept of epileptic and non-epileptic “myoclonus”114–116 “Myoclonus” is a descriptive term for heterogeneous phenomena such as “sudden brief jerk caused by involuntary muscle activity”, “quick muscle regular or irregular jerks”,” a sudden brief, shock-like muscle contraction arising from the central nervous system”, “abrupt, jerky, involuntary movements unassociated with loss of consciousness”. Clinically myoclonus is divided into physiological, essential, epileptic, and symptomatic. 116 Symptomatic causes are more common and include post-hypoxia, toxic-metabolic disorders, reactions to drugs, storage disease, and neurodegenerative disorders.
For definitions of myoclonus, myoclonic and other seizures see Chapter 7 (page 138).
Erratic or fragmentary myoclonus is the defining seizure type that sometimes may appear immediately after birth. The term erratic is because the myoclonias shift typically from one part of the body to another in a random and asynchronous fashion. Erratic myoclonus affects the face or limbs. It is often restricted in a finger, a toe, the eyebrows, eyelids or lips occurring in the same muscle group and often migrating elsewhere usually in an asynchronous and asymmetrical fashion. Myoclonias are brief, single or repetitive, very frequent and nearly continuous. It is exceptional for a baby with early myoclonic encephalopathy to have mild and infrequent jerks.
Massive usually bisynchronous axial myoclonic jerks may start from the onset of the disease or occur later, often interspersed with erratic myoclonias.
Simple focal seizures, often clinically inconspicuous, manifest with eye deviation or autonomic symptoms such as flushing of the face or apnoea. Focal clonic seizures affect any part of the body. Asymmetric tonic posturing also occurs.
Tonic seizures occur frequently and usually appear in the first month of life. They manifest with truncal tonic contraction which usually also involves the limbs. They occur during wakefulness and sleep.
Genuine tonic epileptic spasms are rare and generally appear later. They usually develop within 2–4 months from the onset of myoclonias, they are solitary or in clusters and are more frequent during alert stages than sleep stages.
Psychomotor development may be abnormal from the onset of seizures or arrests and deteriorates rapidly afterwards. There may be marked truncal hypotonia, limb hypertonia, disconjugate eye movements, dyspnoea, opisthotonic or decerebrate posturing. All patients have bilateral pyramidal signs. Practically, there is no trace of intelligent activity. Patients are unable to follow moving objects with their eyes. One patient developed peripheral neuropathy.110
Aetiology
Early myoclonic encephalopathy is a multi-factorial disease with a high incidence of familial cases.52,113 Some may be due to an autosomal recessive inheritance. Inborn errors of metabolism are the most common causes. These are non-ketotic hyperglycinaemia, propionic aciduria, methylmalonic acidaemia, d-glyceric acidaemia, sulphite and xanthine oxidase deficiency, Menkes disease and Zellweger syndrome and molybdenum co-factor deficiency.113 Metabolic causes explain the high incidence of siblings with this disorder.
A case with a clinical picture of early myoclonic encephalopathy and an atypical suppression–burst pattern had full recovery after administration of pyridoxine.119 Lesional brain abnormalities are rare.
Neuropathological findings when available are not consistent. These include depletion of cortical neurones and astrocytic proliferation, severe multifocal spongy changes in the white matter, peri-vascular concentric bodies, demyelination in cerebral hemispheres, imperfect lamination of the deeper cortical layers and unilateral enlargement of the cerebral hemisphere with astrocytic proliferation.120 No pathological abnormalities have been reported in two cases.110
Pathophysiology
It is apparent from the various and diverse causes of early myoclonic encephalopathy that no one single factor appears to be responsible. Most likely early myoclonic encephalopathy and Ohtahara syndrome are the earliest forms of epileptic encephalopathies as detailed in Chapter 7.
Spreafico et al. 121 proposed a common neuropathological basis irrespective of aetiology. They assumed that numerous large spiny neurones scattered in the white matter along the axons of the cortical gyri represent interstitial cells of neocortical histogenesis that failed to follow their natural programming to die at the end of gestation or soon after birth.121
Diagnostic Procedures
These are the same as those for neonatal seizures, attempting to find an aetiological cause. Brain imaging is usually normal at the onset of the disease but progressive cortical and peri-ventricular atrophy often develops. Asymmetrical enlargement of one hemisphere, dilatation of the corresponding lateral ventricle, cortical and peri-ventricular atrophy and exceptionally malformations of cortical development have been reported.
When considering the relatively high rate of inborn errors of metabolism and mainly non-ketotic hyperglycinaemia a thorough metabolic screening is mandatory. This should include serum levels of amino acids and particularly glycine and glycerol metabolites, organic acids and amino acids in the cerebrospinal fluid.
Electroencephalography
The inter-ictal EEG of early myoclonic encephalopathy is a repetitive suppression–burst pattern without physiological rhythms (Figure 5.4). The bursts of high-amplitude spikes and sharp and slow waves last for 1–5 s and alternate with periods of a flat or almost flat EEG lasting 3–10 s. In most cases the suppression–burst pattern becomes more apparent during deep sleep and may not occur in the EEG of wakefulness.113 The suppression–burst pattern may appear late at 1–5 months of age in some cases and characteristically persists for a prolonged period.122
Erratic myoclonias usually do not have an ictal EEG expression and may follow the bursts.
The suppression–burst pattern evolves to atypical hypsarrhythmia or multifocal spikes and sharp waves 3–4 months from onset of the disease. However, this EEG state of atypical hypsarrhythmia is transient and returns to the suppression–burst pattern, which persists for a long time.
Prognosis
Early myoclonic encephalopathy is one of the most dreadful diseases. More than half of patients die within weeks or months from onset and the others develop permanent severe mental and neurological deficits.
Management
There is no effective treatment. Adrenocorticotropic hormone therapy and anti-epileptic medication (clonazepam, nitrazepam, valproate, phenobarbitone and others) are of no benefit. Patients with non-ketotic hyperglycinaemia may benefit from a reduction in dietary protein and administration of 120 mg/kg of sodium benzoate daily though the outcome is commonly very poor.123
A trial with pyridoxine is justifiable.
Clinical noteDiagnostic tips for early myoclonic encephalopathy
Segmental and erratic myoclonias affecting the face and limbs, usually restricted to a finger, the eyebrows and peri-oral muscles, that is nearly continuous and often shifting from place to place. A persistent EEG suppression–burst pattern.
Ohtahara Syndrome
Ohtahara syndrome52,112,113,124,125 is a rare and devastating form of severe epileptic encephalopathy of very early life.
Demographic Data
Onset is mainly around the first 10 days of life, sometimes intra-uterinely or up to 3 months of age. There may be a slight male predominance. The prevalence and incidence are unknown. There are approximately 100 reported cases but this may be an underestimation as many newborn babies with such a severe disease and early death may escape clinico-EEG diagnosis. According to one report ‘attacks of cerebral spasms occur in 1.5–5 per 1000 newborn post-partum’.126
Clinical Manifestations
Ohtahara syndrome manifests with clinico-EEG features of mainly tonic spasms and suppression–burst EEG patterns that consistently occur in the sleeping and waking states.
Tonic spasms usually consist of a forwards tonic flexion lasting 1–10 s that is singular or in long clusters 10–300 times every 24 h. They may be generalised and symmetrical or lateralised. They occur in both the wake and sleep stages. Less often, one-third of the neonates may have erratic focal motor clonic seizures or hemiconvulsions. Alternating hemiconvulsions or GTCS are exceptional. Myoclonic seizures are rare. Erratic myoclonias are not featured.
Aetiology
The most common cause is malformations of cerebral development such as hemimegalencephaly, porencephaly, Aicardi syndrome, olivary-dentate dysplasia, agenesis of mamillary bodies, linear sebaceous naevus syndrome, cerebral dysgenesis and focal cortical dysplasia.113 Rarely, other lesional brain or metabolic disorders may also be responsible.52,113 There are no familial cases.
ILAE Definition of Ohtahara Syndrome
Ohtahara syndrome or ‘early infantile epileptic encephalopathy with suppression–burst’ is defined by the ILAE Commission as follows.19
This syndrome, as described by Ohtahara et al.,127 is defined by very early onset, within the first few months of life, frequent tonic spasms and a suppression–burst EEG pattern in both the waking and sleeping states. Partial seizures may occur. Myoclonic seizures are rare. The aetiology and underlying pathology are obscure. The prognosis is serious with severe psychomotor retardation and seizure intractability. Often there is evolution to West syndrome at age 4–6 months.19
In neuropathological studies, patients with Ohtahara syndrome had the most severe lesions in comparison with early myoclonic encephalopathy and West syndrome.128
Pathophysiology
Ohtahara syndrome is likely to be the earliest age-related specific epileptic reaction of the developing brain to heterogeneous insults similar to those of other epileptic encephalopathies that occur at a later brain maturity age (Chapter 7). This is supported by the fact that, between 2 and 6 months of age, the clinico-EEG features often change to those of West syndrome and later to Lennox–Gastaut syndrome.
There may be a dysfunction of the catecholaminergic and serotonergic systems that may be responsible for this type of neonatal epileptic encephalopathy.128
Diagnostic Procedures
These are the same as for neonatal seizures, involving attempts at detecting an aetiological cause and possible treatment. Brain imaging usually shows severe abnormalities and malformations of cortical development. Metabolic screening is mandatory if brain imaging is normal.
Electroencephalography
The EEG suppression–burst pattern has a pseudo-rhythmic periodicity. The bursts consist of high-amplitude slow waves intermixed with spikes lasting for 2–6 s. The suppression period of a flat or near flat EEG lasts for 3–5 s. The interval between the onsets of two successive bursts is in the range of 5–10 s. This pattern occurs during both the waking and sleeping stages.
According to Ohtahara et al.129 the pseudo-rhythmic appearance of the suppression–burst pattern during wakefulness and sleep distinguishes Ohtahara syndrome from the periodic type of hypsarrhythmia in which periodicity becomes clear during sleep and from the burst–suppression EEGs of severely abnormal neonates.
The Burst–suppression Pattern in Ohtahara Syndrome versus Early Myoclonic Encephalopathy
There are certain differentiating features of burst–suppression pattern between Ohtahara syndrome and early myoclonic encephalopathy. According to Ohtahara122 the suppression–burst pattern of early myoclonic encephalopathy is accentuated during sleep and often may not occur in the awake state while this is continuous in Ohtahara syndrome.122 Furthermore, the suppression–burst pattern appears at the onset of the disease and disappears within the first 6 months of life in Ohtahara syndrome, whereas in early myoclonic encephalopathy the suppression–burst pattern appears at 1–5 months of age in some cases and characteristically persists for a prolonged period.122
On video–EEG and polymyographic recordings, the suppression–burst pattern is associated with tonic spasms of variable duration concomitant with the burst phase.124 Tonic spasms may also occur with the following EEG features.
- Diffuse desynchronisation with disappearance of suppression–burst activity when tonic spasms cluster in intervals of 5–10 s.
- A pattern in which the suppression–burst pattern becomes more frequent, more diffuse and of higher amplitude compared to the inter-ictal pattern.
Progression of Clinical and Electroencephalogram Features to West Syndrome and Lennox–Gastaut Syndrome
There is an age-related evolution of clinical and EEG patterns from Ohtahara syndrome, first to West syndrome and then to Lennox–Gastaut syndrome.
In EEG, a characteristic development is the gradual disappearance of the suppression–burst pattern and the emergence of hypsarrhythmia within 3–6 months from onset. This may again progress later to the slow spike-wave EEG patterns of Lennox–Gastaut syndrome.
The suppression–burst transformation to hypsarrhythmia starts with the gradual emergence of higher amplitude rhythms in the suppression phase, followed by disappearance of the suppression–burst pattern in the awake stage EEG and finally in the sleep stage EEG.129 Changing from hypsarrhythmia to diffuse slow spike-wave, the hypsarrhythmic patterns gradually become fragmented and disappear. They are replaced initially in the awake and subsequently in the sleep EEG by the slow spike-wave patterns of Lennox-Gastaut syndrome.129
Some survivors may show highly localised or entirely unilateral spikes and these patients may frequently have severe focal seizures. Multifocal spikes are frequent while an EEG void of spikes is rather exceptional. Asymmetrical suppression–burst patterns are more likely to develop spike foci and less likely to progress to hypsarrhythmia.
Differential Diagnosis
The main differential diagnosis of Ohtahara syndrome is from early myoclonic encephalopathy (Table 5.4).52,130
Table 5.4
Early myoclonic encephalopathy versus Ohtahara syndrome
Prognosis
This is a devastating syndrome associated with high mortality and morbidity. Half of patients die within weeks or months from onset and the others soon develop permanent severe mental and neurological deficits. Psychomotor development relentlessly deteriorates. The babies become inactive with spastic diplegia, hemiplegia, tetraplegia, ataxia or dystonia. In survivors, the clinical and EEG patterns change to that of West syndrome within a few months from onset and this may also change to Lennox–Gastaut syndrome features if patients reach 2–3 years of age.
According to Ohtahara et al.129 patients with suppression–burst patterns evolving to hypsarrhythmia and then to slow spike-wave EEG have the worse prognosis and a high mortality rate. Conversely, those who develop spike foci, have fewer seizures and less mortality despite severe psychomotor handicaps.
Management
There is no effective treatment. Adrenocorticotropic hormone therapy and anti-epileptic medication of any type are of no benefit. An excellent response to zonisamide was reported in a single case.131 A short-lived beneficial effect of vigabatrin has been reported. Newer drugs have not been tested. Neurosurgery in focal cerebral dysplasia is sometimes beneficial.132
Clinical noteDiagnostic tips for Ohtahara syndrome
Tonic seizures during the awake and sleep stages in the early days or weeks of life are nearly pathognomonic of Ohtahara syndrome. The EEG has a burst–suppression pattern with a pseudo-rhythmic appearance occurring during the awake and sleep stages.
Non-Epileptic Movement Disorders in Neonates and Infants Imitating Seizures
Non-epileptic movement disorders in neonates and infants may be misdiagnosed as epileptic seizures. The commoner of these are:
- Benign neonatal sleep myoclonus
- Benign non-epileptic myoclonus of early infancy (benign non-epileptic infantile spasms)
- Hyperekplexia (familial startle disease)
Benign Neonatal Sleep Myoclonus
Benign neonatal sleep myoclonus133–136 is a common non-epileptic condition misdiagnosed as epileptic seizures and even as infantile spasms.
Demographic Data
Onset is from the first day to 3 weeks of life with a peak at the seventh day. Boys and girls are equally affected. Though common the exact prevalence is unknown.
Clinical Manifestations
The myoclonus occurs during non-REM sleep in otherwise normal neonates. It mainly affects the distal parts of the upper extremities. The lower limbs and axial muscles are less often involved. The myoclonic jerks, synchronous or asynchronous, unilateral or bilateral, mild or violent, usually last for 10–20 s. Occasionally they may occur in repetitive clusters of 2–3 s for 30 min or longer imitating myoclonic status epilepticus or a series of epileptic fits. The myoclonic jerks may get worse with gentle restraint. They abruptly stop when the child is awakened. Sleep is not disturbed.
There are no other clinical manifestations like those accompanying neonatal seizures such as apnoea, autonomic disturbances, automatisms, eye deviation, oral–buccal–lingual movements or crying.
Neurological mental state and development are normal.
Aetiology
This is unknown and the condition does not appear to be familial. The myoclonus is likely to be generated in the brain stem.
Diagnostic Procedures
The diagnosis is based on clinical features. All relevant laboratory studies including sleep EEG during the myoclonus are normal.
Differential Diagnosis
Benign neonatal sleep myoclonus should be easy to differentiate from relevant epileptic disorders in this age group by its occurrence in normal neonates and only during sleep. When in doubt, a normal sleep EEG during the myoclonus is confirmatory of this non-epileptic condition.
Prognosis
The prognosis is excellent with the myoclonus commonly remitting by the age of 2–7 months.
Treatment
There is no need for any treatment though minute doses of clonazepam before bed are often beneficial. Other anti-epileptic drugs are contraindicated.
Benign Non-Epileptic Myoclonus of Early Infancy
Benign Non-Epileptic Infantile Spasms
Benign non-epileptic myoclonus of early infancy,137–140 as described by Fejerman and Lombroso,137 is a paroxysmal, non-epileptic movement disorder of otherwise healthy infants who have normal EEG and development. Its synonym of ‘benign non-epileptic infantile spasms’ is descriptively more accurate than myoclonus.141 It is probably the same disease as ‘shuddering attacks’.142
Demographic Data
Onset is from around 4–12 months of age. Both sexes are probably equally affected.
Clinical Manifestations
The attacks are sudden and brief symmetrical axial flexor spasms mainly of the trunk and often the head. Less frequently, there may be flexion, abduction or adduction of the elbows and knees and extension or elevation of the arms. The spasms do not involve localised muscle groups and there are no focal or lateralising features. Clinically, each spasm lasts for 1–2 s.
The attacks are more likely to occur in clusters, sometimes recurring at frequent and brief intervals several times a day. The intensity of the spasms varies. They are usually mild and inconspicuous but may at times become severe and imitate infantile spasms. They occur in the awake state and are also elicited by excitement, fear, anger, frustration or the need to move the bowels or to void.
Aetiology
This is unknown. Benign non-epileptic infantile spasms may result from an exaggeration of physiological myoclonus.139
Diagnostic Procedures
The diagnosis is based on clinical features. All relevant laboratory studies including sleep and awake stage EEGs during the spasms are normal.
Differential Diagnosis
The main differential diagnosis is from epileptic spasms that may share similar clinical features. A normal ictal and inter-ictal EEG in benign non-epileptic infantile spasms is of decisive significance.
Prognosis
Benign non-epileptic myoclonus of early infancy has a good prognosis with spontaneous remission in the first 5 years of life, usually by age 2–3 years.
Treatment
There is no convincing evidence of any beneficial treatment. Anti-epileptic drugs are unnecessary and potentially harmful.139
Hyperekplexia
Familial Startle Disease
Hyperekplexia or familial startle disease143–149 is the first human disorder shown to result from mutations within a neurotransmitter gene.
Demographic Data
Onset is from intra-uterine life or birth or later at any time from the neonatal period to adulthood. Both sexes are equally affected. It is a rare disorder. Only approximately 150 cases have been reported.
Clinical Manifestations
Clinically hyperekplexia is characterised by:
- pathological and excessive startle responses to unexpected auditory or tactile stimuli (sudden noise, movement or touch)
- severe generalised stiffness (hypertonia in flexion which disappears in sleep).
The startle response is characterised by sudden generalised muscular rigidity and resistance to habituation. In babies the muscle stiffening often causes respiratory impairment and apnoea that may be fatal. In older patients the startle response causes frequent falls, like a log, without loss of consciousness.
If an unborn baby is affected the mother may first notice abnormal intra-uterine movements. In newborn neonates apnoea and sluggish feeding efforts occur as a consequence of episodic extreme stiffening during the first 24 h of life. After the first 24 h surviving infants exhibit the hyperekplexic startle response to nose tapping, which is a useful diagnostic test (page 112).
Clinical phenotypic expression varies from mild to very severe forms.150,151
The minor forms manifest with excessive startle responses only, which are often mild and inconsistent. In infancy these are facilitated by febrile illness whereas in adults these are facilitated by emotional stress.
In the major forms affected neonates occasionally have fatal hypertonia and startle responses result in falls that may be traumatic. There is no impairment of consciousness, but the patient remains temporarily stiff after the attack.
Sleep episodic shaking of the limbs (nocturnal or sleep myoclonus) resembling generalised clonus or repetitive myoclonus is often prominent, lasting for minutes with no impairment of consciousness. The jerks are spontaneous arousal reactions.152
Neurologically, there is generalised muscle hypertonia–stiffness hence the term stiff baby syndrome (which is probably the same disease is hyperekplexia).153 Gait may be unstable, insecure and puppet like. Brain stem and tendon reflexes are exaggerated.
Umbilical and inguinal hernias, presumably due to increased intra-abdominal pressure, are common.
Aetiology
Hyperekplexia is usually inherited as an autosomal dominant and less often recessive trait. It is due to mutations within the GLRA1 gene in chromosome 5q33-35, encoding the alpha 1 subunit of the glycine receptor.147,148,154–157 The minor form of hyperekplexia is seldom due to a genetic defect in the GLRA1 gene.151
Diagnostic Procedures
The nose tap test is the most useful test. Tapping the tip of the nose of an unaffected baby will elicit a blink response or no response, but in hyperekplexia there is an obvious startle response, which is repeated each time the nose is tapped.
The EEG of startle responses in hyperekplexia is normal.158 Slowing of background activity with eventual flattening may occur, but this corresponds to the phase of apnoea, bradycardia and cyanosis.158
Differential Diagnosis
Hyperekplexia in the neonatal period may be misdiagnosed as congenital stiff-man syndrome, startle epilepsy, myoclonic seizures, neonatal tetany, cerebral palsy and drug (phenothiazine) toxicity. Accurate recognition of hyperekplexia in a newborn is important so as to initiate early and appropriate treatment, which may be life saving.
Prognosis
This is generally good in treated patients. Untreated infants experience recurring apnoea until 1 year of age. The exaggerated startle response persists to adulthood. Hypertonia diminished during the course of the first and second year of life and tone is usually almost normal by the age of 3 years. Hypertonia may recur in adult life.
Treatment
There is a dramatic response to clonazepam (0.1–0.2 mg/kg/day).144 A simple manoeuvre to terminate the startle response is forcibly flexing the baby by pressing the head towards the knees. This may be life saving when prolonged stiffness impedes respiration. Affected families are advised to seek genetic counselling.
References
- 1.
- Volpe JJ. Neonatal seizures. Philadelphia: W B Saunders; 1995.
- 2.
- Lombroso CT. Neonatal seizures: historic note and present controversies. Epilepsia. 1996;37(Suppl 3):5–13. [PubMed: 8681914]
- 3.
- Mizrahi EM. Treatment of neonatal seizures. In: Engel J, Pedley TA, editors. Epilepsy. A comprehensive textbook. Lippincott-Raven; 1997. pp. 1295–303.
- 4.
- Mizrahi EM, Plouin P, Kellaway P. Neonatal seizures. In: Engel J, Pedley TA, editors. Epilepsy. A comprehensive textbook. Lippincott-Raven; 1997. pp. 647–63.
- 5.
- Rennie JM. Neonatal seizures. Eur J Pediatr. 1997;156:83–7. [PubMed: 9039506]
- 6.
- Ronen GM, Penney S, Andrews W. The epidemiology of clinical neonatal seizures in Newfoundland: a population-based study. J Pediatr. 1999;134:71–5. [PubMed: 9880452]
- 7.
- Schmid R, Tandon P, Stafstrom CE, Holmes GL. Effects of neonatal seizures on subsequent seizure-induced brain injury. Neurology. 1999;53:1754–61. [PubMed: 10563624]
- 8.
- Watanabe K, Miura K, Natsume J, Hayakawa F, Furune S, Okumura A. Epilepsies of neonatal onset: seizure type and evolution. Dev Med Child Neurol. 1999;41:318–22. [PubMed: 10378757]
- 9.
- Levene M. The clinical conundrum of neonatal seizures. Arch Dis Child Fetal Neonatal Ed. 2002;86:F75–F77. [PMC free article: PMC1721380] [PubMed: 11882546]
- 10.
- Mizrahi EM, Kellaway P. Diagnosis and management of neonatal seizures. Hagerstown: Lippincott Williams & Wilkins; 1999.
- 11.
- Hill A. Neonatal seizures. Pediatr Rev. 2000;21:117–21. [PubMed: 10756174]
- 12.
- Mizrahi EM, Kellaway P. Neonatal seizures. In: Pellock JM, Dodson WE, Bourgeois BFD, editors. Pediatric epilepsy. New York: Demos; 2001. pp. 145–61.
- 13.
- Holmes GL, Khazipov R, Ben Ari Y. New concepts in neonatal seizures. Neuroreport. 2002;13:A3–A8. [PubMed: 11924904]
- 14.
- Scher MS. Controversies regarding neonatal seizure recognition. Epileptic Disord. 2002;4:139–58. [PubMed: 12105077]
- 15.
- Mizrahi E, Watanabe K. Symptomatic neonatal seizures. In: Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. 3. London: John Libbey & Co Ltd; 2002. pp. 15–31.
- 16.
- Tharp BR. Neonatal seizures and syndromes. Epilepsia. 2002;43(Suppl 3):2–10. [PubMed: 12060001]
- 17.
- Mizrahi EM. Seizures in the neonate. In: Wallace SJ, Farrell K, editors. Epilepsy in childen. London: Arnold; 2004. pp. 111–22.
- 18.
- Arzimanoglou A, Guerrini R, Aicardi J. Neonatal seizures. In: Arzimanoglou A, Guerrini R, Aicardi J, editors. Aicardi’s epilepsy in children. Philadelphia: Lippincott Williams & Wilkins; 2004. pp. 188–209.
- 19.
- Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia. 1989;30:389–99. [PubMed: 2502382]
- 20.
- Volpe JJ. Neonatal seizures: current concepts and revised classification. Pediatrics. 1989;84:422–8. [PubMed: 2671912]
- 21.
- Watanabe K, Hara K, Miyazaki S, Hakamada S, Kuroyanagi M. Apneic seizures in the newborn. Am J Dis Child. 1982;136:980–4. [PubMed: 7124705]
- 22.
- Scher MS. Pathologic myoclonus of the newborn: electrographic and clinical correlations. Pediatr Neurol. 1985;1:342–8. [PubMed: 3939749]
- 23.
- Fenichel GM, Olson BJ, Fitzpatrick JE. Heart rate changes in convulsive and nonconvulsive neonatal apnea. Ann Neurol. 1980;7:577–82. [PubMed: 7436362]
- 24.
- Scher MS. Seizures in the newborn infant. Diagnosis, treatment, and outcome. Clin Perinatol. 1997;24:735–72. [PubMed: 9395861]
- 25.
- Perlman JM, Volpe JJ. Seizures in the preterm infant: effects on cerebral blood flow velocity, intracranial pressure, and arterial blood pressure. J Pediatr. 1983;102:288–93. [PubMed: 6822940]
- 26.
- Watanabe K, Hara K, Hakamada S, Negoro T, Sugiura M, Matsumoto A, et al. Seizures with apnea in children. Pediatrics. 1982;70:87–90. [PubMed: 7088639]
- 27.
- Mizrahi EM, Kellaway P. Characterization and classification of neonatal seizures. Neurology. 1987;37:1837–44. [PubMed: 3683874]
- 28.
- Minchom P, Niswander K, Chalmers I, Dauncey M, Newcombe R, Elbourne D, et al. Antecedents and outcome of very early neonatal seizures in infants born at or after term. Br J Obstet Gynaecol. 1987;94:431–9. [PubMed: 3580326]
- 29.
- Nelson KB, Lynch JK. Stroke in newborn infants. Lancet Neurol. 2004;3:150–8. [PubMed: 14980530]
- 30.
- Aicardi J. Neonatal seizures. In: Dam M, Gram L, editors. Comprehensive epileptology. New York: Raven Press; 1991. pp. 99–112.
- 31.
- Aicardi J. Epilepsy in Children. New York: Raven Press; 1994.
- 32.
- Moshe SL. Seizures in the developing brain. Neurology. 1993;43:S3–S7. [PubMed: 8232986]
- 33.
- Velisek L, Moshe SL. Pathophysiology of seizures and epilepsy in the immature brain:Cells, synapses and circuits. In: Pellock JM, Dodson WE, Bourgeois BFD, editors. Pediatric epilepsy. New York: Demos; 2001. pp. 1–23.
- 34.
- Holmes GL, Ben Ari Y. Seizures in the developing brain: perhaps not so benign after all. Neuron. 1998;21:1231–4. [PubMed: 9883716]
- 35.
- Jensen FE. Acute and chronic effects of seizures in the developing brain: experimental models. Epilepsia. 1999;40(Suppl 1):S51–S58. [PubMed: 10421561]
- 36.
- Mizrahi EM. Acute and chronic effects of seizures in the developing brain: lessons from clinical experience. Epilepsia. 1999;40(Suppl 1):S42–S50. [PubMed: 10421560]
- 37.
- Kuzniecky RI. Neuroimaging in pediatric epileptology. In: Pellock JM, Dodson WE, Bourgeois BFD, editors. Pediatric epilepsy. New York: Demos; 2001. pp. 133–43.
- 38.
- Aso K, Abdab-Barmada M, Scher MS. EEG and the neuropathology in premature neonates with intraventricular hemorrhage. J Clin Neurophysiol. 1993;10:304–13. [PubMed: 8408597]
- 39.
- Holmes GL, Lombroso CT. Prognostic value of background patterns in the neonatal EEG. J Clin Neurophysiol. 1993;10:323–52. [PubMed: 8408599]
- 40.
- Scher MS. Neonatal encephalopathies as classified by EEG-sleep criteria: severity and timing based on clinical/pathologic correlations. Pediatr Neurol. 1994;11:189–200. [PubMed: 7880332]
- 41.
- Clancy RR. The contribution of EEG to the understanding of neonatal seizures. Epilepsia. 1996;37(Suppl 1):S52–S59. [PubMed: 8647052]
- 42.
- Ortibus EL, Sum JM, Hahn JS. Predictive value of EEG for outcome and epilepsy following neonatal seizures. Electroencephalogr Clin Neurophysiol. 1996;98:175–85. [PubMed: 8631277]
- 43.
- Marret S, Parain D, Menard JF, Blanc T, Devaux AM, Ensel P, et al. Prognostic value of neonatal electroencephalography in premature newborns less than 33 weeks of gestational age. Electroencephalogr Clin Neurophysiol. 1997;102:178–85. [PubMed: 9129573]
- 44.
- de Weerd AW, Despland PA, Plouin P, Neonatal EEG. The International Federation of Clinical Neurophysiology. Electroencephalogr Clin Neurophysiol Suppl. 1999;52:149–57. [PubMed: 10590984]
- 45.
- Patrizi S, Holmes GL, Orzalesi M, Allemand F. Neonatal seizures: characteristics of EEG ictal activity in preterm and fullterm infants. Brain Dev. 2003;25:427–37. [PubMed: 12907278]
- 46.
- Bednarek N. Video-EEG monitoring in neonates: indications. Epileptic Disord. 2001.:SI21–SI24. 3 Spec No 2. [PubMed: 11827843]
- 47.
- Scher MS, Aso K, Beggarly ME, Hamid MY, Steppe DA, Painter MJ. Electrographic seizures in preterm and full-term neonates: clinical correlates, associated brain lesions, and risk for neurologic sequelae. Pediatrics. 1993;91:128–34. [PubMed: 8416475]
- 48.
- Mizrahi EM, Tharp BR. A characteristic EEG pattern in neonatal herpes simplex encephalitis. Neurology. 1982;32:1215–20. [PubMed: 6890154]
- 49.
- Arzimanoglou A, Guerrini R, Aicardi J. Aicardi’s epilepsy in children. Philadelphia: Lippincott Williams & Wilkins; 2004.
- 50.
- Lombroso CT. Early myoclonic encephalopathy, early infantile epileptic encephalopathy, and benign and severe infantile myoclonic epilepsies: a critical review and personal contributions. J Clin Neurophysiol. 1990;7:380–408. [PubMed: 2120281]
- 51.
- Tendero GA, Lopez MV, Arcas MJ, Roche Herrero MC, Martinez BA. Neonatal EEG trace of burst suppression. Etiological and evolutionary factors. Rev Neurol. 2001;33:514–8. [PubMed: 11727229]
- 52.
- Aicardi J, Ohtahara S. Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, editors. Severe neonatal epilepsies with suppression-burst pattern. London: John Libbey & Co Ltd; Epileptic syndromes in infancy, childhood and adolescence. (3) 2002:33–44.
- 53.
- Kellaway P, Mizrahi EM. Neonatal seizures. In: Luders H, Lesser RP, editors. Epilepsy. Electroclinical syndromes. Berlin: Springer-Verlag; 1987. pp. 13–47.
- 54.
- Mizrahi EM. Cleve Clin J Med. Suppl Pt 1. Vol. 56. 1989. Clinical and neurophysiologic correlates of neonatal seizures; pp. S100–S104. [PubMed: 2655986]
- 55.
- Scher MS. Stimulus-evoked electrographic patterns in neonates: an abnormal form of reactivity. Electroencephalogr Clin Neurophysiol. 1997;103:679–91. [PubMed: 9546495]
- 56.
- Clancy RR, Legido A, Lewis D. Occult neonatal seizures. Epilepsia. 1988;29:256–61. [PubMed: 3371282]
- 57.
- Weiner SP, Painter MJ, Geva D, Guthrie RD, Scher MS. Neonatal seizures: electroclinical dissociation. Pediatr Neurol. 1991;7:363–8. [PubMed: 1764139]
- 58.
- Biagioni E, Ferrari F, Boldrini A, Roversi MF, Cioni G. Electroclinical correlation in neonatal seizures. Europ J Paediatr Neurol. 1998;2:117–25. [PubMed: 10726833]
- 59.
- Boylan GB, Panerai RB, Rennie JM, Evans DH, Rabe-Hesketh S, Binnie CD. Cerebral blood flow velocity during neonatal seizures. Arch Dis Child Fetal Neonatal Ed. 1999;80:F105–F110. [PMC free article: PMC1720914] [PubMed: 10325785]
- 60.
- Garcias Da Silva LF, Nunes ML, da Costa JC. Risk factors for developing epilepsy after neonatal seizures. Pediatr Neurol. 2004;30:271–7. [PubMed: 15087106]
- 61.
- Rennie JM, Boylan GB. Neonatal seizures and their treatment. Curr Opin Neurol. 2003;16:177–81. [PubMed: 12644746]
- 62.
- Fischer JH, Lockman LA, Zaske D, Kriel R. Phenobarbital maintenance dose requirements in treating neonatal seizures. Neurology. 1981;31:1042–4. [PubMed: 7196518]
- 63.
- Donn SM, Grasela TH, Goldstein GW. Safety of a higher loading dose of phenobarbital in the term newborn. Pediatrics. 1985;75:1061–4. [PubMed: 4000780]
- 64.
- Painter MJ, Scher MS, Stein AD, Armatti S, Wang Z, Gardiner JC, et al. Phenobarbital compared with phenytoin for the treatment of neonatal seizures. N Engl J Med. 1999;341:485–9. [PubMed: 10441604]
- 65.
- Maytal J, Novak GP, King KC. Lorazepam in the treatment of refractory neonatal seizures. J Child Neurol. 1991;6:319–23. [PubMed: 1940133]
- 66.
- Sheth RD, Buckley DJ, Gutierrez AR, Gingold M, Bodensteiner JB, Penney S. Midazolam in the treatment of refractory neonatal seizures. Clin Neuropharmacol. 1996;19:165–70. [PubMed: 8777770]
- 67.
- Boylan GB, Rennie JM, Chorley G, Pressler RM, Fox GF, Farrer K, et al. Second-line anticonvulsant treatment of neonatal seizures: a video-EEG monitoring study. Neurology. 2004;62:486–8. [PubMed: 14872039]
- 68.
- Wallin A, Nergardh A, Hynning PA. Lidocaine treatment of neonatal convulsions, a therapeutic dilemma. Eur J Clin Pharmacol. 1989;36:583–6. [PubMed: 2776817]
- 69.
- Singh B, Singh PHI, Khan M, Majeed-Saidan M. Treatment of neonatal seizures with carbamazepine. J Child Neurol. 1996;11:378–82. [PubMed: 8877605]
- 70.
- Baxter PS, Gardner-Medwin D, Barwick DD, Ince P, Livingston J, Murdoch-Eaton D. Vigabatrin monotherapy in resistant neonatal seizures. Seizure. 1995;4:57–9. [PubMed: 7788110]
- 71.
- Barr PA, Buettiker VE, Antony JH. Efficacy of lamotrigine in refractory neonatal seizures. Pediatr Neurol. 1999;20:161–3. [PubMed: 10082350]
- 72.
- Massingale TW, Buttross S. Survey of treatment practices for neonatal seizures. J Perinatol. 1993;13:107–10. [PubMed: 8515301]
- 73.
- Brod SA, Ment LR, Ehrenkranz RA, Bridgers S. Predictors of success for drug discontinuation following neonatal seizures. Pediatr Neurol. 1988;4:13–7. [PubMed: 3233103]
- 74.
- Ellison PH, Horn JL, Franklin S, Jones MG. The results of checking a scoring system for neonatal seizures. Neuropediatrics. 1986;17:152–7. [PubMed: 2429227]
- 75.
- Engel J Jr. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: Report of the ILAE Task Force on Classification and Terminology. Epilepsia. 2001;42:796–803. [PubMed: 11422340]
- 76.
- Plouin P. Benign familial neonatal convulsions and benign idiopathic neonatal convulsions. In: Engel J jr, Pedley TA, editors. Epilepsy. A comprehensive textbook. Lippincott-Raven; 1997. pp. 2247–55.
- 77.
- Hirsch E, Velez A, Sellal F, Maton B, Grinspan A, Malafosse A, et al. Electroclinical signs of benign neonatal familial convulsions. Ann Neurol. 1993;34:835–41. [PubMed: 8250533]
- 78.
- Hirsch E, Saint-Martin A, Marescaux C. Benign familial neonatal convulsions: a model of idiopathic epilepsy. Rev Neurol (Paris). 1999;155:463–7. [PubMed: 10472660]
- 79.
- Plouin P, Anderson VE. Benign familial and non-familial neonatal seizures. In: Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. 3. London: John Libbey & Co Ltd; 2002. pp. 3–13.
- 80.
- Vidaurre JA, Ballaban-Gil KR, Plouin P, Moshe SL. Benign neonatal familial seizures. In: Gilman S, editor. Medlink Neurology. San Diego SA: Arbor Publishing Corp; 2004.
- 81.
- Zonana J, Silvey K, Strimling B. Familial neonatal and infantile seizures: an autosomal-dominant disorder. Am J Med Genet. 1984;18:455–9. [PubMed: 6476007]
- 82.
- Miles DK, Holmes GL. Benign neonatal seizures. J Clin Neurophysiol. 1990;7:369–79. [PubMed: 2211994]
- 83.
- Ronen GM, Rosales TO, Connolly M, Anderson VE, Leppert M. Seizure characteristics in chromosome 20 benign familial neonatal convulsions. Neurology. 1993;43:1355–60. [PubMed: 8327138]
- 84.
- Ryan SG, Wiznitzer M, Hollman C, Torres MC, Szekeresova M, Schneider, et al. Benign familial neonatal convulsions: evidence for clinical and genetic heterogeneity. Ann Neurol. 1991;29:469–73. [PubMed: 1859177]
- 85.
- Leppert M, Anderson VE, Quattlebaum T, Stauffer D, O’Connell P, Nakamura Y, et al. Benign familial neonatal convulsions linked to genetic markers on chromosome 20. Nature. 1989;337:647–8. [PubMed: 2918897]
- 86.
- Lewis TB, Leach RJ, Ward K, O’Connell P, Ryan SG. Genetic heterogeneity in benign familial neonatal convulsions: identification of a new locus on chromosome 8q. Am J Hum Genet. 1993;53:670–5. [PMC free article: PMC1682419] [PubMed: 8102508]
- 87.
- Singh NA, Charlier C, Stauffer D, DuPont BR, Leach RJ, Melis R, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nature Genetics. 1998;18:25–9. [PubMed: 9425895]
- 88.
- Biervert C, Steinlein OK. Structural and mutational analysis of KCNQ2, the major gene locus for benign familial neonatal convulsions. Hum Genet. 1999;104:234–40. [PubMed: 10323247]
- 89.
- Lee WL, Biervert C, Hallmann K, Tay A, Dean JC, Steinlein OK. A KCNQ2 splice site mutation causing benign neonatal convulsions in a Scottish family. Neuropediatrics. 2000;31:9–12. [PubMed: 10774989]
- 90.
- Hirose S, Zenri F, Akiyoshi H, Fukuma G, Iwata H, Inoue T, et al. A novel mutation of KCNQ3 (c.925T—>C) in a Japanese family with benign familial neonatal convulsions. Ann Neurol. 2000;47:822–6. [PubMed: 10852552]
- 91.
- Castaldo P, del Giudice EM, Coppola G, Pascotto A, Annunziato L, Taglialatela M. Benign familial neonatal convulsions caused by altered gating of KCNQ2/KCNQ3 potassium channels. J Neurosci. 2002;22:RC199. [PMC free article: PMC6758678] [PubMed: 11784811]
- 92.
- Leppert M, Singh N. Benign familial neonatal epilepsy with mutations in two potassium channel genes. Curr Opin Neurol. 1999;12:143–7. [PubMed: 10226745]
- 93.
- Singh NA, Westenskow P, Charlier C, Pappas C, Leslie J, Dillon J, et al. KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain. 2003;126:2726–37. [PubMed: 14534157]
- 94.
- Berkovic SF, Heron SE, Giordano L, Marini C, Guerrini R, Kaplan RE, et al. Benign familial neonatal-infantile seizures: Characterization of a new sodium channelopathy. Ann Neurol. 2004;55:550–7. [PubMed: 15048894]
- 95.
- Heron SE, Crossland KM, Andermann E, Phillips HA, Hall AJ, Bleasel A, et al. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet. 2002;360:851–2. [PubMed: 12243921]
- 96.
- Okada M, Wada K, Kamata A, Murakami T, Zhu G, Kaneko S. Impaired M-current and neuronal excitability. Epilepsia. 2002;43(Suppl 9):36–8. [PubMed: 12383278]
- 97.
- Aso K, Watanabe K. Benign familial neonatal convulsions: generalized epilepsy? Pediat Neurol. 1992;8:226–8. [PubMed: 1622522]
- 98.
- Baxter P, Kandler R. Benign familial neonatal convulsions: abnormal intrauterine movements, provocation by feeding and ICTAL EEG. Seizure. 1997;6:485–6. [PubMed: 9530946]
- 99.
- Bye AM. Neonate with benign familial neonatal convulsions: recorded generalized and focal seizures. Pediatr Neurol. 1994;10:164–5. [PubMed: 8024668]
- 100.
- Shevell MI, Sinclair DB, Metrakos K. Benign familial neonatal seizures: clinical and electroencephalographic characteristics. Pediatr Neurol. 1986;2:272–5. [PubMed: 3508699]
- 101.
- Pettit RE, Fenichel GM. Benign familial neonatal seizures. Arch Neurol. 1980;37:47–8. [PubMed: 7350900]
- 102.
- Kaplan RE, Lacey DJ. Benign familial neonatal-infantile seizures. Am J Med Genet. 1983;16:595–9. [PubMed: 6660252]
- 103.
- Psenka TM, Holden KR. Benign familial neonatal convulsions; psychosocial adjustment to the threat of recurrent seizures. Seizure. 1996;5:243–5. [PubMed: 8902929]
- 104.
- Dehan M, Quilleron D, Navelet Y, et al. Les convulsions du cinquieme jour de vie: un nouveau syndrome? Arch Fr Pediatr. 1977;37:730–42. [PubMed: 931532]
- 105.
- Plouin P. Benign neonatal convulsions (familial and non-familial). In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. London: John Libbey & Company; 1992. pp. 2–11.
- 106.
- Ballaban KR, Plouin P, Moshe SL. Benign neonatal familial seizures. In: Gilman S, editor. Medlink Neurology. San Diego SA: Arbor Publishing Corp; 2004.
- 107.
- Vidaurre JA, Ballaban-Gil KR, Plouin P, Moshe SL. Benign neonatal seizures (nonfamilial). In: Gilman S, editor. Medlink Neurology. San Diego SA: Arbor Publishing Corp; 2004.
- 108.
- Sperber EF, Haas KZ, Stanton PK, Moshe SL. Resistance of the immature hippocampus to seizure-induced synaptic reorganization. Brain Res Dev. 1991;60:88–93. [PubMed: 1717181]
- 109.
- North KN, Storey GN, Henderson-Smart DJ. Fifth day fits in the newborn. Aust Paediatr J. 1989;25:284–7. [PubMed: 2590128]
- 110.
- Dalla Bernardina B, Dulac O, Fejerman N, Dravet C, Capovilla G, Bondavalli S, et al. Early myoclonic epileptic encephalopathy (E.M.E.E.). Eur J Pediatr. 1983;140:248–52. [PubMed: 6414818]
- 111.
- Bruel H, Boulloche J, Chabrolle JP, Layet V, Poinsot J. Early myoclonic epileptic encephalopathy and non-ketotic hyperglycemia in the same family. Arch Pediatr. 1998;5:397–9. [PubMed: 9759159]
- 112.
- Ohtahara S, Yamatogi Y. Epileptic encephalopathies in early infancy with suppression-burst. J. Clin. Neurophysiol. 2003;20:398–407. [PubMed: 14734930]
- 113.
- Ohtahara S, Yamatogi Y, Ohtsuka Y. Early epileptic encephalopathies. In: Wallace SJ, Farrell K, editors. Epilepsy in childen. London: Arnold; 2004. pp. 133–41.
- 114.
- Agarwal P, Frucht SJ. Myoclonus. Curr Opin Neurol. 2003;16:515–21. [PubMed: 12869812]
- 115.
- Faught E. Clinical presentations and phenomenology of myoclonus. Epilepsia. 2003;44(Suppl 11):7–12. [PubMed: 14641566]
- 116.
- Caviness JN, Brown P. Myoclonus: current concepts and recent advances. Lancet Neurol. 2004;3:598–607. [PubMed: 15380156]
- 117.
- Blume WT, Luders HO, Mizrahi E, Tassinari C, van Emde BW, Engel J Jr. Glossary of descriptive terminology for ictal semiology: report of the ILAE task force on classification and terminology. Epilepsia. 2001;42:1212–8. [PubMed: 11580774]
- 118.
- Janz D, Inoue Y, Seino M. Myoclonic seizures. In: Engel JJ, Pedley TA, editors. Epilepsy: A comprehensive Textbook. Philadelphia: Lippincott-Raven Publishers; 1997. pp. 591–603.
- 119.
- Wang PJ, Lee WT, Hwu WL, Young C, Yau KI, Shen YZ. The controversy regarding diagnostic criteria for early myoclonic encephalopathy. Brain Dev. 1998;20:530–5. [PubMed: 9840674]
- 120.
- Aicardi J. Early myoclonic encephalopathy (neonatal myoclonic encephalopathy). In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. London: John Libbey & Company; 1992. pp. 13–23.
- 121.
- Spreafico R, Angelini L, Binelli S, Granata T, Rumi V, Rosti D, et al. Burst suppression and impairment of neocortical ontogenesis: electroclinical and neuropathologic findings in two infants with early myoclonic encephalopathy. Epilepsia. 1993;34:800–8. [PubMed: 8404728]
- 122.
- Ohtahara S, Ohtsuka Y, Oka E. Epileptic encephalopathies in early infancy. Indian J Pediatr. 1997;64:603–12. [PubMed: 10771894]
- 123.
- Chien YH, Hsu CC, Huang A, Chou SP, Lu FL, Lee WT, et al. Poor outcome for neonatal-type nonketotic hyperglycinemia treated with high-dose sodium benzoate and dextromethorphan. J Child Neurol. 2004;19:39–42. [PubMed: 15032382]
- 124.
- Fusco L, Pachatz C, Di Capua M, Vigevano F. Video/EEG aspects of early-infantile epileptic encephalopathy with suppression-bursts (Ohtahara syndrome). Brain Dev. 2001;23:708–14. [PubMed: 11701283]
- 125.
- Yamatogi Y, Ohtahara S. Early-infantile epileptic encephalopathy with suppression-bursts, Ohtahara syndrome; its overview referring to our 16 cases. Brain Dev. 2002;24:13–23. [PubMed: 11751020]
- 126.
- Romann D, Golz N, Garbe W. [Ohtahara syndrome] Geburtshilfe Frauenheilkd. 1996;56:393–5. [PubMed: 8964455]
- 127.
- Ohtahara S, Ishida T, Oka E, Yamatogy Y, Inoue H. On the specific age-dependent epileptic syndromes: the early-infantile epileptic encephalopathy with suppression-burst. No To Hattatsu. 1976;8:270–80.
- 128.
- Itoh M, Hanaoka S, Sasaki M, Ohama E, Takashima S. Neuropathology of early-infantile epileptic encephalopathy with suppression-bursts; comparison with those of early myoclonic encephalopathy and West syndrome. Brain Dev. 2001;23:721–6. [PubMed: 11701285]
- 129.
- Ohtahara S, Ohtsuka Y, Yamatogi Y, Oka E, Inoue H. Early-infantile epileptic encephalopathy with suppression-bursts. In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. London: John Libbey & Company; 1992. pp. 25–34.
- 130.
- Murakami N, Ohtsuka Y, Ohtahara S. Early infantile epileptic syndromes with suppression-bursts: early myoclonic encephalopathy vs. Ohtahara syndrome. Jpn J Psychiatry Neurol. 1993;47:197–200. [PubMed: 8271543]
- 131.
- Ohno M, Shimotsuji Y, Abe J, Shimada M, Tamiya H. Zonisamide treatment of early infantile epileptic encephalopathy. Pediatr Neurol. 2000;23:341–4. [PubMed: 11068168]
- 132.
- Komaki H, Sugai K, Maehara T, Shimizu H. Surgical treatment of early-infantile epileptic encephalopathy with suppression-bursts associated with focal cortical dysplasia. Brain Dev. 2001;23:727–31. [PubMed: 11701286]
- 133.
- Di Capua M, Fusco L, Ricci S, Vigevano F. Benign neonatal sleep myoclonus: clinical features and video- polygraphic recordings. Mov Disord. 1993;8:191–4. [PubMed: 8474488]
- 134.
- Daoust-Roy J, Seshia SS. Benign neonatal sleep myoclonus. A differential diagnosis of neonatal seizures. Am J Dis Child. 1992;146:1236–41. [PubMed: 1415056]
- 135.
- Caraballo R, Yepez I, Cersosimo R, Fejerman N. Benign neonatal sleep myoclonus. Rev Neurol. 1998;26:540–4. [PubMed: 9796000]
- 136.
- Sheldon SH. Benign neonatal sleep myoclonus. In: Gilman S, editor. Medlink Neurology. San Diego SA: Arbor Publishing Corp; 2004.
- 137.
- Lombroso CT, Fejerman N. Benign myoclonus of early infancy. Ann Neurol. 1977;1:138–43. [PubMed: 889296]
- 138.
- Pachatz C, Fusco L, Vigevano F. Benign myoclonus of early infancy. Epilep Disord. 1999;1:57–61. [PubMed: 10937134]
- 139.
- Maydell BV, Berenson F, Rothner AD, Wyllie E, Kotagal P. Benign myoclonus of early infancy: an imitator of West’s syndrome. J Child Neurol. 2001;16:109–12. [PubMed: 11292215]
- 140.
- Wilner AN, Ballaban KR, Moshe SL. Benign nonepileptic infantile spasms. In: Gilman S, editor. Medlink Neurology. San Diego SA: Arbor Publishing Corp; 2004.
- 141.
- Dravet C, Giraud N, Bureau M, Roger J, Gobbi G, Dalla Bernardina B. Benign myoclonus of early infancy or benign non-epileptic infantile spasms. Neuropediatrics. 1986;17:33–8. [PubMed: 3960282]
- 142.
- Kanazawa O. Shuddering attacks-report of four children. Pediatr Neurol. 2000;23:421–4. [PubMed: 11118798]
- 143.
- Andermann F, Keene DL, Andermann E, Quesney LF. Startle disease or hyperekplexia: further delineation of the syndrome. Brain. 1980;103:985–97. [PubMed: 6777025]
- 144.
- Ryan SG, Sherman SL, Terry JC, Sparkes RS, Torres MC, Mackey RW. Startle disease, or hyperekplexia: response to clonazepam and assignment of the gene (STHE) to chromosome 5q by linkage analysis. Ann Neurol. 1992;31:663–8. [PubMed: 1355335]
- 145.
- Tijssen MA, Schoemaker HC, Edelbroek PJ, Roos RA, Cohen AF, van Dijk JG. The effects of clonazepam and vigabatrin in hyperekplexia. J Neurol Sci. 1997;149:63–7. [PubMed: 9168167]
- 146.
- Vergouwe MN, Tijssen MA, Peters AC, Wielaard R, Frants RR. Hyperekplexia phenotype due to compound heterozygosity for GLRA1 gene mutations. Ann Neurol. 1999;46:634–8. [PubMed: 10514101]
- 147.
- Saul B, Kuner T, Sobetzko D, Brune W, Hanefeld F, Meinck HM, et al. Novel GLRA1 missense mutation (P250T) in dominant hyperekplexia defines an intracellular determinant of glycine receptor channel gating. J Neurosci. 1999;19:869–77. [PMC free article: PMC6782149] [PubMed: 9920650]
- 148.
- Shiang R, Ryan SG, Zhu YZ, Fielder TJ, Allen RJ, Fryer A, et al. Mutational analysis of familial and sporadic hyperekplexia. Ann Neurol. 1995;38:85–91. [PubMed: 7611730]
- 149.
- Praveen V, Patole SK, Whitehall JS. Hyperekplexia in neonates. Postgrad Med J. 2001;77:570–2. [PMC free article: PMC1757896] [PubMed: 11524514]
- 150.
- Bernasconi A, Regli F, Schorderet DF, Pescia G. Familial hyperekplexia: startle disease. Clinical, electrophysiological and genetic study of a family. Rev Neurol (Paris). 1996;152:447–50. [PubMed: 8944241]
- 151.
- Tijssen MA, Vergouwe MN, van Dijk JG, Rees M, Frants RR, Brown P. Major and minor form of hereditary hyperekplexia. Mov Disord. 2002;17:826–30. [PubMed: 12210885]
- 152.
- de Groen JH, Kamphuisen HA. Periodic nocturnal myoclonus in a patient with hyperexplexia (startle disease). J Neurol Sci. 1978;38:207–13. [PubMed: 213539]
- 153.
- Cioni G, Biagioni E, Bottai P, Castellacci AM, Paolicelli PB. Hyperekplexia and stiff-baby syndrome: an identical neurological disorder? Ital J Neurol Sci. 1993;14:145–52. [PubMed: 8509269]
- 154.
- Findlay GS, Phelan R, Roberts MT, Homanics GE, Bergeson SE, Lopreato GF, et al. Glycine receptor knock-in mice and hyperekplexia-like phenotypes: comparisons with the null mutant. J Neurosci. 2003;23:8051–9. [PMC free article: PMC6740502] [PubMed: 12954867]
- 155.
- Miraglia DG, Coppola G, Bellini G, Ledaal P, Hertz JM, Pascotto A. A novel mutation (R218Q) at the boundary between the N-terminal and the first transmembrane domain of the glycine receptor in a case of sporadic hyperekplexia. J Med Genet. 2003;40:e71. [PMC free article: PMC1735464] [PubMed: 12746425]
- 156.
- Gomeza J, Ohno K, Hulsmann S, Armsen W, Eulenburg V, Richter DW, et al. Deletion of the mouse glycine transporter 2 results in a hyperekplexia phenotype and postnatal lethality. Neuron. 2003;40:797–806. [PubMed: 14622583]
- 157.
- Castaldo P, Stefanoni P, Miceli F, Coppola G, del Giudice EM, Bellini G, et al. A novel hyperekplexia-causing mutation in the pre-transmembrane segment 1 of the human glycine receptor alpha1 subunit reduces membrane expression and impairs gating by agonists. J Biol Chem. 2004;279:25598–604. [PubMed: 15066993]
- 158.
- Brown P. Neurophysiology of the startle syndrome and hyperekplexia. Adv Neurol. 2002;89:153–9. [PubMed: 11968441]
Footnotes
- *
Note on a recent development
‘Benign familial neonatal–infantile seizures’ is a newly described clinically intermediate variant between benign familial neonatal seizures and benign familial infantile seizures. The disorder can now be strongly suspected clinically and the families can be given an excellent prognosis.94,95
- Neonatal Seizures
- Neonatal Syndromes
- Benign Familial Neonatal Seizures
- Benign Neonatal Seizures (Non-Familial)
- Early Myoclonic Encephalopathy
- Ohtahara Syndrome
- Non-Epileptic Movement Disorders in Neonates and Infants Imitating Seizures
- Benign Neonatal Sleep Myoclonus
- Benign Non-Epileptic Myoclonus of Early Infancy
- Hyperekplexia
- References
- Neonatal Seizures and Neonatal Syndromes - The EpilepsiesNeonatal Seizures and Neonatal Syndromes - The Epilepsies
- Homo sapiens solute carrier family 12 member 4 (SLC12A4), transcript variant 3, ...Homo sapiens solute carrier family 12 member 4 (SLC12A4), transcript variant 3, mRNAgi|225579062|ref|NM_001145962.1|Nucleotide
Your browsing activity is empty.
Activity recording is turned off.
See more...