Molecular Pathogenesis
KCNA1 encodes the potassium voltage-gated channel subfamily A member 1, commonly known as the α-subunit of the voltage-gated delayed-rectifier potassium channel Kv1.1. Voltage-gated potassium channels (Kv) play key roles in neurotransmission and nerve cell physiology. They shorten the duration of action potentials, modulate the release of neurotransmitters, and control the excitability, electrical properties, and firing pattern of central and peripheral neurons [Pessia 2004]. In particular, Kv1.1 channels regulate neuromuscular transmission and control the release of β-aminobutyric acid (GABA) from cerebellar basket cells onto Purkinje cells [Herson et al 2003]. The Kv1.1 channel opens upon membrane depolarization, after which potassium flow results in a hyperpolarizing effect that is necessary to limit neuronal excitability [Pessia 2004]. The channel is known for its role in controlling the excitability of cerebellar, hippocampal, cortical, and peripheral nervous system neurons [Brunetti et al 2012, D'Adamo et al 2015a].
Functional studies have shown that pathogenic missense variants in KCNA1 (the only gene currently known to be associated with EA1) result in loss of function of the channel. Kv1.1 channel function in EA1 is impaired by altering the channel's gating kinetics, voltage dependence, assembly, and trafficking [Imbrici et al 2006, Hasan et al 2017; for a review see D'Adamo et al 2015b].
Homomeric Kv1.1 channels are tetrameric structures composed of four identical α-subunit monomers. Each monomer is encoded by KCNA1. However, potassium channel diversity is greatly enhanced by the ability of Kv1.1 to co-assemble with α-subunits of other members of the Kv1 family to form heterotetrameric channels with properties different from the parental homomeric channels. Kv1.1 is mostly found co-assembled with Kv1.2 subunits; they are expressed together at cerebellar basket cell terminals and at the juxtaparanodal region of motor axons. A pathogenic variant in Kv1.1 affects the function of the Kv1.1/1.2 heteromeric channel to which they contribute [Hasan et al 2017]. D'Adamo et al first demonstrated that proteins encoded by KCNA1 pathogenic variants associated with EA1 alter the expression and gating properties of heteromeric channels composed of human Kv1.2 and Kv1.1 subunits [D'Adamo et al 1999, Rea et al 2002].
Kv1.1 channels possess a slow process of inactivation, which has been named C-type or P-type depending on the structural determinants of this process that have been located within the C-terminus and pore region. Kv channels may also exhibit fast N-type inactivation that is caused by a "ball-and-chain" mechanism of pore occlusion. Fast inactivation may be conferred to non-inactivating channels by auxiliary subunits such as Kvβ1.1 and Kvβ1.2. Four β subunits participate in the ion channel complex and provide four inactivation particles; notable example: channels composed of Kv1.1, Kv1.4, and Kvβ1.1 subunits that are expressed in hippocampal mossy fiber boutons [Geiger & Jonas 2000]. Proteins encoded by KCNA1 pathogenic variants also impair the function of hetero-oligomeric complexes comprising Kv1.1, Kv1.4, and Kvβ1.x subunits in distinct ways [Imbrici et al 2006, Imbrici et al 2011]. These studies raised the question as to whether other allelic variations, whose gene products may or may not form hetero-oligomeric complexes with Kv1.1 subunits, may underlie a similar channelopathy.
Gene structure.
KCNA1 has a transcript of 7,983 nucleotides with a coding region of 1,488. There are two exons, but the coding region is located entirely within exon 2. The reference sequences in Table 5 include the correction of a sequence error (see Table 5, footnote 1). For a detailed summary of gene and protein information, see Table A, Gene.
Benign variants. In 5% of control chromosomes analyzed by Zuberi et al [1999], two silent changes in the coding sequence were observed.
Pathogenic variants. To date, more than 30 KCNA1 pathogenic variants have been identified by sequence analysis (see ). Most are missense variants that are distributed throughout the gene; however, nonsense variants and small deletions have also been identified [Eunson et al 2000, Shook et al 2008].
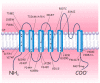
Schematic drawing of the conventional membrane topology of a human Kv1.1 subunit. Four such subunits comprise a functional homotetrameric channel. Different subunits belonging to the Kv1 subfamily may form heterotetrameric channels. The positions of pathogenic (more...)
Interestingly, four different variants of the highly conserved threonine 226 residue, located within the second transmembrane segment, have been identified [Rajakulendran et al 2007]. In particular, the amino acid change p.Thr226Arg is associated with epilepsy, infantile contractures, postural abnormalities, and skeletal deformities. Although the defects caused by the p.Thr226Ala, p.Thr226Arg, and p.Thr226Met amino acid changes on channel functions are virtually identical, they lead to diverse phenotypes.
Table 5.
KCNA1 Pathogenic Variants Discussed in This GeneReview
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.676A>G | p.Thr226Ala | NM_000217.2 1
NP_000208.2 |
| c.677C>G | p.Thr226Arg |
| c.677C>T | p.Thr226Met |
| c.1223T>C | p.Val408Ala |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Normal gene product.
KCNA1 encodes the voltage-gated K+ channel Kv1.1. The predicted 496-amino-acid Kv1.1 protein contains six hydrophobic segments with the N- and C-termini residing inside the cell. The S4 segment of each Kv1.1 subunit comprises the main voltage sensor that opens the channel by undergoing a conformational rearrangement on membrane depolarization. The S5-S6 loop (H5 region) contributes to the ion-conducting pore. The GYG residues, residing within this loop, control the K+ selectivity of the channel.
Abnormal gene product. The molecular mechanisms underlying episodic ataxia type 1 have been established by determining the functional properties of wild type and several mutant channels in Xenopus oocytes or mammalian cell lines [Adelman et al 1995, D'Adamo et al 1998, Zerr et al 1998, D'Adamo et al 1999, Zuberi et al 1999, Eunson et al 2000, Manganas et al 2001, Imbrici et al 2003, Cusimano et al 2004, Imbrici et al 2006, Imbrici et al 2007, Imbrici et al 2008, Imbrici et al 2009, Imbrici et al 2011, D'Adamo et al 2015a]. Overall, these studies have shown that allelic variations underlying EA1 impair channel function and reduce the outward K+ flux through the channel, although with highly variable effects on aspects of channel expression and gating.
Regarding channel gating, KCNA1 pathogenic variants may alter the protein structure and affect the kinetics of opening and closing, voltage dependence, and N- and C-type inactivation [D'Adamo et al 1998, D'Adamo et al 1999, Maylie et al 2002, Imbrici et al 2006, Imbrici et al 2009, Imbrici et al 2011, D'Adamo et al 2015b, Hasan et al 2017].
Individuals with EA1 are heterozygous for a KCNA1 pathogenic variant, possessing a normal and a mutated allele, which may be equally expressed. Therefore, channels composed of wild type and mutated subunits may be formed. Co-expression systems, which mimic the heterozygous condition, have shown that some mutated subunits exert dominant negative effects on wild type subunits, resulting in less than half the normal current, whereas others have virtually no effect on surface expression. It has been shown that KCNA1 allelic variations also alter the function of heteromeric channels containing different subunits, demonstrating that pathogenic variants in a single gene disrupt the functions of other closely related proteins [D'Adamo et al 1999, Rea et al 2002, Imbrici et al 2006, Hasan et al 2017]. Based on these findings, a model accounting for the cerebellar symptoms of EA1 was proposed by D'Adamo and colleagues (see ).
Proposed effects of EA1-causing pathogenic variants on basket cell and Purkinje cell inhibitory outputs The diagram shows a basket cell that has synapses on the initial segment and soma of a number of Purkinje cells from the cerebellar cortex of an unaffected (more...)
A mouse model of EA1 has been generated by introducing a pathogenic variant analogous to the human p.Val408Ala EA1 pathogenic variant into the murine ortholog, Kcna1. These animals showed impaired motor performance and altered cerebellar GABAergic transmission from the basket cells to the Purkinje cells [Herson et al 2003]. Such Kv1.1 knock-in ataxic mice also exhibited spontaneous myokymic activity exacerbated by fatigue, ischemia, and low temperature [Brunetti et al 2012]. Spontaneous myokymic discharges were present despite motor nerve axotomy, suggesting that the motor nerve is an important generator of myokymic activity. This study also showed that altered Ca2+ homeostasis in motor axons of mutated animals may contribute to spontaneous myokymic activity [Brunetti et al 2012].
The causes that trigger the paroxysms of ataxia remain elusive, although a phenomenon akin to spreading acidification of the cerebellar cortex has been suggested [Chen et al 2005].