Clinical Description
To date, approximately 150 individuals with a clinical diagnosis of Cantú syndrome have been identified; a pathogenic variant in ABCC9 or KCNJ8 has been identified in 107 individuals [Grange et al 2019]. The following description of the phenotypic features associated with this condition is based on this report.
Table 2.
Cantú Syndrome: Frequency of Select Features
View in own window
| Feature | % of Persons
w/Feature | Comment |
|---|
| Polyhydramnios | 57% | |
| Prematurity (<37 weeks) | 58% | |
| Neonatal hypertrichosis | 99% | |
| Macrosomia | 38% | Birth weight >4,000 g |
| Generalized edema at birth | 43% | |
| Macrocephaly | 48% of adults
studied | |
| Skeletal abnormalities | 19% | Usually asymptomatic so detection dependent on imaging; not all persons had full skeletal survey |
| Gastroesophageal reflux | 42% | |
| Cardiovascular findings | PDA | 58% | |
| Valvular defects | 18% | Bicuspid aortic valve, mitral valve regurgitation, aortic valve stenosis |
| Cardiac enlargement | 64% | |
| Dilated aortic root | 32% | |
| Pericardial effusion | 25% | |
| Tortuous vascularity | 100% (10/10) on neurovascular imaging | True number is unknown in entire population; neuroimaging at Washington University in St Louis showed that all tested persons have this finding in head & neck. |
| Pulmonary hypertension | 24% | Seen in infancy; typically resolves w/age |
| Peripheral edema | 51% | Usually develops in teenagers & young adults |
| Developmental delays | 63% | Present in infants & young children related to hypotonia, but improve over time; most have normal intellect. |
| Hypotonia | 65% | |
| Headaches | 40% | Often migraine-type headache w/assoc symptoms |
| Seizures | 24% | Various types |
| Behavioral issues 1 | ADHD | 19% | |
| ASD | 16% | |
| OCD | 13% | |
| Anxiety | 13% | |
| Depression | 19% | |
ADHD = attention-deficit/hyperactivity disorder; ASD = autism spectrum disorder; OCD = obsessive-compulsive disorder; PDA = patent ductus arteriosus
- 1.
Self-reported in many individuals
Prenatal. Many pregnancies with a fetus with Cantú syndrome are complicated by polyhydramnios, leading in some instances to repeated amniotic fluid reductions as well as preterm labor and delivery.
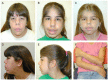
Newborns. All newborns with Cantú syndrome have hypertrichosis with thick scalp hair and excessive hair growth on the forehead, face, back, and extremities. Some have thick and/or curly eyelashes. The hypertrichosis usually persists over time.
Many newborns have macrosomia (large birth weight and birth length) and macrocephaly.
Generalized edema at birth (observed on occasion) usually resolves spontaneously.
Growth. Ultimate adult height is usually within the normal range; however, short stature has been seen in a few individuals.
Macrocephaly, often present at birth, typically persists throughout life. Some individuals who do not have macrocephaly at birth have developed progressive macrocephaly in childhood.
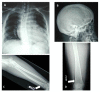
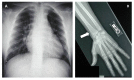
Skeletal abnormalities are usually asymptomatic and identified on radiographs. Characteristic skeletal abnormalities have included thickening of the calvaria, broad ribs, platyspondyly, ovoid vertebral bodies, scoliosis, narrow thorax and shoulders, pectus carinatum, hypoplastic ischium and pubic bones, Erlenmeyer-flask-like long bones with metaphyseal flaring, narrow obturator foramen, and coxa vara.
Gastroesophageal reflux is reported by just under half of individuals. A smaller percentage report intestinal dysfunction characterized by chronic constipation or slow intestinal motility.
Cardiac findings include the following:
Cardiac enlargement, with increased ventricular mass but normal chamber wall thickness and enlarged chambers of the heart, often present at birth. Despite the enlarged cardiac chambers, cardiac function is typically normal and ventricular contractility is increased on imaging studies [
Grange et al 2006,
Grange et al 2019]. Some patients note exercise intolerance, but others have been able to participate in organized sports without difficulty.
Patent ductus arteriosus (PDA) in 58% (and described as extremely large in some), often requiring surgical closure in infancy or early childhood
Bicuspid aortic valve with and without stenosis
Pericardial effusion in about 25% of affected individuals. Small pericardial effusions may be asymptomatic; large fluid accumulations result in symptoms such as exercise intolerance and require intervention.
Dilated aortic root and ascending aorta are present in about two thirds of individuals. The natural history is poorly understood. However, development of an aortic aneurysm is rare. Aortic aneurysm requiring surgical intervention was reported in one individual with an
ABCC9 pathogenic variant [
Hiraki et al 2014].
Tortuous vascularity including tortuous retinal vessels and multiple tortuous pulmonary arteriovenous communications have been reported [Scurr et al 2011, Grange et al 2019]. Abnormal tortuous vasculature in the brain is present in essentially all individuals who have been imaged specifically [Leon Guerrero et al 2016, Grange et al 2019].
Pulmonary hypertension has been reported in infants and young children, although the natural history is not well understood [Cantú et al 1982, Robertson et al 1999, Lazalde et al 2000, Kobayashi et al 2010, Scurr et al 2011]. In one child, pulmonary hypertension secondary to partial pulmonary venous obstruction was associated with severe mitral valve regurgitation that spontaneously resolved by age eight years [Kobayashi et al 2010]. In another individual, progressive (and ultimately fatal) pulmonary hypertension was reported [Park et al 2014]. In the majority of cases, pulmonary hypertension is mild and improves with age [Grange et al 2019].
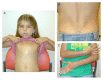
Generalized edema, which may be present at birth, spontaneously resolves. Subsequently, edema involving the lower extremities and occasionally the arms and hands may develop over time, usually in adolescence or early adulthood. Puffiness of the eyelids is often observed. In one individual, lymphangiography demonstrated dilated lymphatic vessels in the legs with delayed lymphatic drainage [García-Cruz et al 2011]. In contrast, lymphatic studies were normal in another individual [Scurr et al 2011]. Therefore, it is unclear at this time whether the observed swelling is edema or lymphedema.
Intellect. Although the majority of affected individuals have normal intellect, mild learning disabilities and/or developmental delays have been observed, including delay in acquisition of early motor milestones (most likely related to decreased muscle tone) and delay in speech development. Ultimately, most affected individuals attend regular schools, and some are described as having a high IQ [Scurr et al 2011, Grange et al 2019].
Seizures are reported in about one quarter of individuals. Febrile, tonic-clonic, and absence seizure types have been observed as well as temporal lobe epilepsy.
Headaches are reported by many individuals, especially migraine-type headaches with associated aura, photophobia, and phonophobia, and occasionally with transient hemiparesis.
Behavioral problems have been reported in some individuals, including anxiety, mood swings, obsessive-compulsive disorder, and tics [Scurr et al 2011, Grange et al 2019]. Attention-deficit/hyperactivity disorder, autism spectrum disorder, and depression may also be present. Many individuals have self-reported these issues.
Features of a connective tissue abnormality are observed in many individuals with Cantú syndrome, including wrinkled or loose skin especially at birth, deep palmar and plantar creases, and joint hyperextensibility. Some have decreased subcutaneous fat with the appearance of a muscular build in childhood.
Less frequent features
Umbilical hernia
Pyloric stenosis
Ptosis
Craniosynostosis involving the sagittal and coronal sutures in one individual [
Hiraki et al 2014]
The three individuals reported thus far with a pathogenic variant in KCNJ8 had typical clinical features seen in Cantú syndrome [Brownstein et al 2013, Cooper et al 2014, Chihara et al 2020]. The individual reported by Brownstein et al [2013] had the following additional abnormalities:
Brain MRI: cerebral atrophy and thin corpus callosum
Multiple tortuous venous collaterals and lack of flow in the inferior sagittal sinus
Systemic vasculature: dilated hepatic and celiac arteries, dilated and tortuous intrahepatic arteries and veins