

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Mobley HLT, Mendz GL, Hazell SL, editors. Helicobacter pylori: Physiology and Genetics. Washington (DC): ASM Press; 2001.

Both sequence diversity and population structures can vary widely among pathogenic bacterial species. In some species, all isolates are highly similar, whereas in other species, any two unrelated isolates can be easily distinguished by simple fingerprinting methods. Strains of Helicobacter pylori have been known since the late 1980s to exhibit considerable genetic diversity (24, 28). However, systematic analyses of the genetic diversity and population structure of H. pylori have only begun recently. The first part of this chapter contains a general introduction to bacterial population genetics, and the second part reviews the available data on the genetic diversity within H. pylori and the mechanisms that have generated this diversity.
Bacterial Population Genetics
Bacterial Population Structures
Three types of population structures were described by Maynard Smith and colleagues (30): clonal, panmictic, and epidemic. In clonal species, genetic differences between individual isolates reflect changes that have accumulated sequentially since the descent from ancestral bacteria. Genetic diversity arises by intragenomic genetic events including point mutations, deletions, insertions, and rearrangements. Genetic information from members of the current population can allow the reconstruction of a parsimonious evolutionary history, and the accumulated genetic diversity can be depicted as a tree. The neutral mutation theory (21) predicts that selectively neutral sequence polymorphisms at synonymous sites are transient and will either be eradicated or fixed within the population by random drift after a time period that correlates with the effective population size. Periodic selection and selective sweeps (27) also purge bacterial populations of sequence polymorphisms. Geographical separation can result in different polymorphisms and fixed mutations in distinct areas. Geographical spread is accompanied by bottlenecks and define eliminate sequence variation within a population and can speed fixation of individual polymorphisms (32).
The sequence polymorphism within a clonal species can be used to estimate the time that has passed since the existence of the population's last common ancestor, the "molecular Eve" (15). In contrast, a lack of sequence divergence in a species or population indicates that insufficient time has elapsed since the existence of its last common ancestor (or the last selective sweep) for divergence to accumulate. For example, based on the lack of sequence diversity, it has been calculated that the last common ancestor of Mycobacterium tuberculosis existed 12,000 years ago (47) and that Yersinia pestis evolved from Yersinia pseudotuberculosis 2,000 to 15,000 years ago (4). These bacteria show little sequence diversity because they are not only clonal but also young. Similar arguments may also apply to Bacillus anthracis, which also shows very little sequence diversity (18).
Other bacteria are clonal, although they have existed long enough to accumulate sequence diversity. For example, Salmonella enterica infects mammals and reptiles and is thought to be derived from an ancestor that existed before mammals and reptiles evolved about 120 million years ago (37). Sequences of many genes from S. enterica yield similar phylogenetic trees (46), indicating clonal descent.
Panmictic species
Clonal population structures can only exist if recombination and horizontal genetic exchange are rare. Horizontal genetic exchange can occur via transformation, conjugation, and transduction. This may lead to the integration of foreign DNA that may originate from other members of the same species (intraspecies recombination) or from closely or distantly related species, including bacteria and bacteriophages (interspecies recombination). Recombination disrupts the clonal structure because the newly introduced DNA does not share the same evolutionary history as the other genes within the genome. If recombination is frequent, phylogenetic trees will differ depending on the locus under analysis and alleles at different loci will be in linkage equilibrium. The resulting population structure is called panmictic, as exemplified by Neisseria gonorrhoeae (30).
Epidemic population structure
If particular clones within a recombining species expand and spread very rapidly (e.g., because they colonize a naive host population), they may transiently possess a clonal population structure. One example is the epidemic clones of Neisseria meningitidis. Transformation with both homologous and heterologous DNA occurs frequently within N. meningitidis, resulting in panmixis. However, most serogroup A isolates and several other hypervirulent clonal groupings show an apparently clonal population structure over a period of decades (26, 30, 32).
Genetic Heterogeneity of H. pylori
Historical Data
As early as 1986, it was recognized that H. pylori exhibits an unusual degree of genetic heterogeneity. Langenberg et al. showed that the HindIII restriction patterns of H. pylori chromosomal DNAs from 16 unrelated patients were unique although multiple isolates from one patient shared a common pattern (24). These observations were confirmed and extended by Majewski and Goodwin, who showed that 69 isolates from Australia and one from the United Kingdom each possessed a unique HindIII restriction pattern (28). Majewski and Goodwin also presented evidence for mixed infections of humans with multiple H. pylori strains. Similar conclusions were obtained with other study populations by various high-resolution typing methods, including ribotyping, restriction fragment length polymorphism (RFLP), and random amplified polymorphic DNA (RAPD) analysis (6, 7, 39). Isolates with similar or identical fingerprints have been found within families (9, 36, 57) and in an institution containing children with encephalopathy (55).
Nucleotide Sequence Variability in H. pylori
The nucleotide sequence diversity of H. pylori exceeds that of many other bacterial species. Furthermore, almost every nucleotide sequence from unrelated isolates is unique, an unprecedented situation. Kansau et al. (17) found that the nucleotide sequence of a 294-bp fragment of the glmM (formerly ureC) gene differed for 29 strains of H. pylori. Similar results have since been described for other genes and from larger samples. For example, 54 flaB flagellin gene sequences from German isolates and 33 flaA and vacA sequences from Canadian strains were all unique (49). However, identical flaB sequences were found in three pairs of strains among 22 isolates from the Cape Coloured population in South Africa, suggesting that the pool of different alleles may be smaller in some geographic regions than in Europe and Canada (49). Similarly, two of the Cape Coloured isolates possessed identical vacA sequences, and two others possessed identical flaA sequences.
High allelic diversity could in theory reflect high mutation rates or frequent recombination, or a combination of both. Although recombination does not create polymorphic sites, it can increase the number of unique sequences by shuffling polymorphic nucleotides and might even import nucleotide diversity from other species.
The long-term frequency with which polymorphic nucleotides arise in a species is reflected by the mean frequency of synonymous nucleotide changes per potential synonymous site (DS). For H. pylori, mean DS values have been calculated for several virulence-associated and housekeeping genes (2). The mean DS values from housekeeping genes were also compared to those from other pathogenic species (1, 49) and were found to be higher than for all species tested except N. meningitidis (Fig. 1). The reason for the elevated frequency of sequence polymorphism in H. pylori and N. meningitidis is still unclear. Wang et al. (56) have suggested that the elevated sequence diversity in H. pylori reflects a high mutation rate, possibly due to mutator phenotypes caused by the lack of genes involved in DNA repair or reduced activity of those genes. Similar explanations have also been invoked for O157:H7 strains of Escherichia coli (25). Yet, the distance to S. enterica serovar Typhimurium is the same for O157:H7 and K-12 strains of E. coli, indicating that mutator phenotypes have not had any long-term effects on evolution (58). While it is conceivable that some H. pylori strains may have a mutator phenotype, and that such strains generate frequent mutations, there is currently no direct evidence that mutators play any role in the evolution of this pathogen. Furthermore, the observed differences in sequence diversity between different bacterial species can readily be explained by differences of population structure and population size (3). At equilibrium, the sequence diversity of a population is directly related to the effective population size. Sequence diversity is reduced by purification events such as bottlenecks, founder effects, and selective sweeps that remove genotypic diversity from the population. The high frequency of polymorphic nucleotides in H. pylori may reflect a large effective population size combined with rare purification events. Indeed, H. pylori lacks a synonymous codon usage bias, indicating that periods of competitive exponential growth are rare (23).
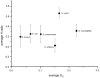
Figure 1
Comparison of the mean homoplasy ratio (H) and mean genetic distance at synonymous sites (DS) among different bacterial species. The sources of the data for these analyses are given in reference . Modified from reference with permission.
Evidence for Recombination in H. pylori
Several sources of data provided indirect evidence for a panmictic population structure of H. pylori with frequent recombination. These include mosaicism in the vacA vacuolating cytotoxin gene (8), linkage equilibrium of housekeeping genes (as assayed by multilocus enzyme electrophoresis) (14), and linkage equilibrium of restriction length polymorphisms (43).
In many bacterial species, recombination results in mosaic genes that can be recognized by visual inspection of sequences. The statistical significance of such mosaic structures can be determined by computational methods developed by Sawyer (44) and Stephens (48), and by Maynard Smith's maximum chi-square test (30). However, these tests can fail when recombination is too frequent (29), and indeed none of these tests yielded unequivocal results when applied to flaB, vacA, and flaA sequences from H. pylori (Suerbaum and Achtman, unpublished). These tests also do not readily quantitate the frequency of recombination. In contrast, the homoplasy test, described by Maynard and Smith in 1998 (31), detects recombination even when mosaics have been obscured due to very high frequencies and provides quantitative estimates of the impact of recombination on the population structure.
The homoplasy test calculates the sequence diversity and the number of homoplasies among informative, synonymous polymorphic sites within a set of homologous sequences. Homoplasies are polymorphic nucleotides that occur in multiple, independent branches of a phylogenetic tree. They can arise by repeated independent mutations or by recombination. The homoplasy test therefore calculates the expected frequency of homoplasies in a maximal parsimony tree for the observed degree of sequence polymorphism according to both mutation and recombination. It then defines a range of expected frequencies of homoplasies ranging from 0.0 (clonal) to 1.0 (free recombination) and places the observed number of homoplasies on this range. The result is the homoplasy ratio H, which estimates the frequency of recombinants within the test set of sequences. The flaB and flaA flagellin genes and a fragment of the vacA gene from H. pylori strains isolated in Germany and Canada yielded homoplasy ratios of 0.8 to 0.9, indicating that individual polymorphic sites were essentially in linkage equilibrium (49). Sequences of seven housekeeping genes in 20 strains isolated from 12 countries also yielded very high homoplasy ratios (2). These homoplasy ratios are much higher than those for housekeeping genes from other bacterial species (Fig. 1), indicating that among bacterial species that have been analyzed, recombinants are most frequent in H. pylori. An independent quantitative measure of the frequency of recombination events is yielded by compatibility matrices that give a visual impression of the degree of clonality and can reveal reticulate evolution that is caused by homoplasies or recombination (16). Compatibility matrices yielded results that were consistent with frequent recombination in H. pylori (2, 49).
Recombinant sequences can only arise during mixed infections with multiple H. pylori strains and will be lost by purification during long-term colonization and transmission from person to person. The frequency of both recombination and purification remains to be quantitated. However, Kersulyte et al. (19) identified multiple strains with different recombinant genotypes from a single patient in Lithuania.
Geographic Variation in H. pylori
Extensive global communication might be expected to result in rapid dissemination of bacteria that are spread horizontally, and epidemic bacteria can spread globally within short time periods (32). In contrast, pathogens transmitted vertically within families would spread as slowly as human migrations between geographic areas, resulting in biogeographic diversity that reflects the original founder populations that were first introduced into particular areas. Biogeographic differences have been described for the JC virus (5) and Haemophilus influenzae (34) but have not yet been investigated extensively for any bacterial pathogen. Biogeographic diversity might also reflect differences in genetic susceptibility or diet of the humans resident in different areas.
Analyses of the virulence-associated cagA gene showed that different sequence families were found in China and The Netherlands (51). At first, this observation seemed to reflect a feature of cagA because no geographic differences were observed for glmM (formerly ureC). Further analyses of strains collected from different regions have confirmed that East Asian cagA alleles differ from those of strains isolated in Europe, the Americas, Africa, or India, which form a uniform group for this gene (2, 53). Geographical partitioning was also observed for two fragments of vacA, termed the signal sequence (s) and the middle (m) region (52, 54), while a different fragment recombined freely among Canadian isolates (49). vacA m-region alleles that distinguish European/American, East Asian, and Indian families of alleles have also been described (33). Similar geographic partitioning was also observed for hspA, the H. pylori GroES homolog (35).
Biogeographical diversity of three virulence-associated genes such as cagA, vacA, and hspA could arise by the various mechanisms described above, ranging from founder effects associated with vertical transmission to selection for antigenic or functional variants with differential fitness for certain host populations. The vacA m region is involved in the determination of cell-type specificity of the toxin (40). The ability to be tyrosine-phosphorylated differs among variants of CagA (38), although the biological relevance of this observation is not yet known (45). Unlike GroES in other bacterial species, H. pylori hspA is also associated with virulence and can induce protective immunity (50). Thus, for all three loci, biogeographic differences might conceivably be associated with selection due to genetic differences between human populations.
In a recombining population, biogeographical diversity for numerous genes that are not under selection would probably represent founder effects and vertical transmission. Although initial analyses with the glmM housekeeping gene did not yield evidence of geographic partitioning (51), a multilocus sequencing approach with diverse housekeeping genes showed that 20 strains of H. pylori from 12 countries differed according to their sources (2). Six isolates from East Asians differed from isolates from the remainder of the world and were called the "Asian" clone. Three isolates from two Africans and one African American seemed to form a second distinct group ("clone 2"), and the other 11 isolates from Europe, the Americas, and Australia formed a heterogeneous third group. These biogeographical groupings probably reflect founder populations that were physically isolated from each other until recently. Exceptional alleles in the Asian group and in clone 2 that are closer to alleles from the other groupings indicate occasional recombination has happened, but most strains belonging to each group were similar at the majority of the loci tested. Subsequent analyses of the same housekeeping genes from additional isolates from Indonesia and South Africa have confirmed the original conclusions (Achtman and Suerbaum, manuscript in preparation), but further analyses are needed to test whether virulence genes such as cagA also fall into three populations.
Multilocus sequence typing (MLST) was originally developed to study the epidemiology and population genetics of N. meningitidis, Streptococcus pneumoniae, and Staphylococcus aureus (12, 13, 26). In these species, MLST is used to recognize highly related isolates that contain identical alleles at seven distinct loci. MLST in its original form is not applicable to H. pylori, in which almost every strain possesses unique sequences at each locus. However, it has proved to be a very powerful method for analyzing biogeographical differences through phylogenetic analyses of H. pylori sequences and may help elucidate the evolutionary history of this organism.
Do H. pylori Genotypes Reflect Human Migrations?
Biogeographical diversity of H. pylori genotypes might reflect the history of the human hosts (10), and the analysis of larger strain collections might correlate with the history of human migrations. Kersulyte et al. (20) provided initial evidence supporting this hypothesis. Strains from Spaniards, Peruvians, and Guatemalan Ladinos (mixed European-Amerindian ancestry) possessed similar RFLP patterns for a locus from the cag pathogenicity island and differed markedly from East Asian strains. Amerindians migrated from Siberia and were geographically separated from Europeans until several hundred years ago. If their Siberian ancesters carried H. pylori that is related to East Asian isolates, then some alleles related to the East Asian alleles should have been detected. Possibly, the Siberian migrants were not colonized with H. pylori and first acquired these bacteria after the arrival of the Spanish conquistadors (20). However, alternative hypotheses could also explain these data, such as complete replacement of a resident H. pylori population by a Spanish type or a selective sweep of the gene examined through the resident bacteria. The study does, however, demonstrate the potential of using H. pylori allelic variation to investigate human ancestries and migrations.
Microevolution during Chronic Colonization
Long-term carriage of H. influenzae (11) or Pseudomonas aeruginosa (41) is accompanied by progressive genetic changes. An initial study failed to detect genomic changes in paired H. pylori isolates obtained 2 years apart from 20 patients (42), but two subsequent studies have shown that H. pylori strains can indeed change during chronic colonization. RAPD and RFLP analysis showed that the six strains isolated from a single patient belonged to two different types: type A (one isolate) and type B (five isolates) (19). The RAPD patterns of type B were similar but exhibited subtle differences, suggesting that genetic variants of the original strain had arisen during chronic infection. Kersulyte et al. (19) also demonstrated that recombination had occurred between strains of RAPD types A and B on at least six occasions, thus accounting for the observed heterogeneity. In a separate study, 11 pairs of sequential H. pylori isolates isolated 7 to 10 years apart were uniform for all specific markers tested, including nucleotide sequences of two gene fragments (22). However, more or less subtle changes of RAPD or RFLP patterns were detected in all pairs, suggesting that intragenomic changes had occurred. Kuipers et al. (22) also noted that later isolates expressed significantly less Lewisy surface antigen than the earlier isolates. These results indicate that microevolution can occur frequently during long-term colonization within single individuals.
The analysis of numerous H. pylori strains from diverse geographic regions by a unified multilocus sequencing system might reveal fascinating details of the biogeographic diversity of both the bacteria and their human hosts. The work load could well be shared among different laboratories if the same genes were analyzed, and the combined efforts could test whether there are parallels between the evolution of pathogenic bacteria and their human hosts.
Acknowledgments
Work on the population genetics of H. pylori in our laboratories was funded by grants from the Deutsche Forschungsgemeinschaft.
References
- 1.
- Achtman, M. 2001.Population structure of Helicobacter pylori and other pathogenic bacterial species, p. 311–321. In M. Achtman and S. Suerbaum (ed.), Helicobacter pylori: Molecular and Cellular Biology. Horizon Scientific Press, Wymondham, United Kingdom.
- 2.
- Achtman M., Azuma T., Berg D. E., Ito Y., Morelli G., Pan Z. J., Suerbaum S., Thompson S. A., van der Ende A., van Doorn L. J. Recombination and clonal groupings within Helicobacter pylori from different geographic regions. Mol. Microbiol. 1999;32:459–470. [PubMed: 10320570]
- 3.
- Achtman M., Suerbaum S. Sequence variation in Helicobacter pylori. Trends Microbiol. 2000;8:57–58. [PubMed: 10664596]
- 4.
- Achtman M., Zurth K., Morelli G., Torrea G., Carniel E. Yersinia pestis, the cause of pandemic plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. USA. 1999;96:14043–14048. [PMC free article: PMC24187] [PubMed: 10570195]
- 5.
- Agostini H. T., Yanagihara R., Davis V., Ryschkewitsch C. F., Stoner G. L. Asian genotypes of JC virus in Native Americans and in a Pacific Island population: markers of viral evolution and human migration. Proc. Natl. Acad. Sci. USA. 1997;94:14542–14546. [PMC free article: PMC25048] [PubMed: 9405649]
- 6.
- Akopyanz N., Bukanov N. O., Westblom T. U., Berg D. E. PCR-based RFLP analysis of DNA sequence diversity in the gastric pathogen Helicobacter pylori. Nucleic Acids Res. 1992;20:6221–6225. [PMC free article: PMC334508] [PubMed: 1361982]
- 7.
- Akopyanz N., Bukanov N. O., Westblom T. U., Kresovich S., Berg D. E. DNA diversity among clinical isolates of Helicobacter pylori detected by PCR-based RAPD fingerprinting. Nucleic Acids Res. 1992;20:5137–5142. [PMC free article: PMC334296] [PubMed: 1408828]
- 8.
- Atherton J. C., Cao P., Peek R. M., Tummuru M. K. R., Blaser M. J., Cover T. L. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. J. Biol. Chem. 1995;270:17771–17777. [PubMed: 7629077]
- 9.
- Bamford K. B., Bickley J., Collins J. S., Johnston B. T., Potts S., Boston V., Owen R. J., Sloan J. M. Helicobacter pylori: comparison of DNA fingerprints provides evidence for intrafamilial infection. Gut. 1993;34:1348–1350. [PMC free article: PMC1374539] [PubMed: 8244100]
- 10.
- Covacci A., Telford J. L., Del Giudice G., Parsonnet J., Rappuoli R. Helicobacter pylori virulence and genetic geography. Science. 1999;284:1328–1333. [PubMed: 10334982]
- 11.
- Duim B., Vogel L., Puijk W., Jansen H. M., Meloen R. H., Dankert J., van Alphen L. Fine mapping of outer membrane protein P2 antigenic sites which vary during persistent infection by Haemophilus influenzae. Infect. Immun. 1996;64:4673–4679. [PMC free article: PMC174430] [PubMed: 8890224]
- 12.
- Enright M. C., Day N. P., Davies C. E., Peacock S. J., Spratt B. G. Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J. Clin. Microbiol. 2000;38:1008–1015. [PMC free article: PMC86325] [PubMed: 10698988]
- 13.
- Enright M. C., Spratt B. G. A multilocus sequence typing scheme for Streptococcus pneumoniae: identification of clones associated with serious invasive disease. Microbiology. 1998;144:3049–3060. [PubMed: 9846740]
- 14.
- Go M. F., Kapur V., Graham D. Y., Musser J. M. Population genetic analysis of Helicobacter pylori by multilocus enzyme electrophoresis: extensive allelic diversity and recombinational population structure. J. Bacteriol. 1996;178:3934–3938. [PMC free article: PMC232656] [PubMed: 8682800]
- 15.
- Hudson R. R. Gene geneologies and the coalescent process. Oxford Surv. Evol. Biol. 1990;7:1–44.
- 16.
- Jakobsen I. B., Easteal S. A program for calculating and displaying compatibility matrices as an aid in determining reticulate evolution in molecular sequences. Comput. Appl. Biosci. 1996;12:291–295. [PubMed: 8902355]
- 17.
- Kansau I., Raymond J., Bingen E., Courcoux P., Kalach N., Bergeret M., Braimi N., Dupont C., Labigne A. Genotyping of Helicobacter pylori isolates by sequencing of PCR products and comparison with the RAPD technique. Res. Microbiol. 1996;147:661–669. [PubMed: 9157493]
- 18.
- Keim P., Kalif A., Schupp J., Hill K., Travis S. E., Richmond K., Adair D. M., Hugh-Jones M., Kuske C. R., Jackson P. Molecular evolution and diversity in Bacillus anthracis as detected by amplified fragment length polymorphism markers. J. Bacteriol. 1997;179:818–824. [PMC free article: PMC178765] [PubMed: 9006038]
- 19.
- Kersulyte D., Chalkauskas H., Berg D. E. Emergence of recombinant strains of Helicobacter pylori during human infection. Mol. Microbiol. 1999;31:31–43. [PubMed: 9987107]
- 20.
- Kersulyte D., Mukhopadhyay A. K., Velapatino B., Su W., Pan Z., Garcia C., Hernandez V., Valdez Y., Mistry R. S., Gilman R. H., Yuan Y., Gao H., Alarcon T., Lopez-Brea M., Balakrish N. G., Chowdhury A., Datta S., Shirai M., Nakazawa T., Ally R., Segal I., Wong B. C., Lam S. K., Olfat F. O., Boren T., Engstrand L., Torres O., Schneider R., Thomas J. E., Czinn S., Berg D. E. Differences in genotypes of Helicobacter pylori from different human populations. J. Bacteriol. 2000;182:3210–3218. [PMC free article: PMC94509] [PubMed: 10809702]
- 21.
- Kimura, M. 1983. The Neutral Theory of Molecular Evolution. Cambridge University Press, Cambridge, United Kingdom.
- 22.
- Kuipers E. J., Israel D. A., Kusters J. G., Gerrits M. M., Weel J., van der Ende A., van der Hulst R. W., Wirth H. P., Nikanne J., Thompson S. A., Blaser M. J. Quasispecies development of Helicobacter pylori observed in paired isolates obtained years apart from the same host. J. Infect. Dis. 2000;181:273–282. [PMC free article: PMC2766531] [PubMed: 10608776]
- 23.
- Lafay B., Atherton J. C., Sharp P. M. Absence of translationally selected synonymous codon usage bias in Helicobacter pylori. Microbiology. 2000;146:851–860. [PubMed: 10784043]
- 24.
- Langenberg W., Rauws E. A., Widjojokusumo A., Tytgat G. N., Zanen H. C. Identification of Campylobacter pyloridis isolates by restriction endonuclease DNA analysis. J. Clin. Microbiol. 1986;24:414–417. [PMC free article: PMC268925] [PubMed: 3020084]
- 25.
- LeClerc J. E., Li B., Payne W. L., Cebula T. A. High mutation frequencies among Escherichia coli and Salmonella pathogens. Science. 1996;274:1208–1211. [PubMed: 8895473]
- 26.
- Maiden M. C., Bygraves J. A., Feil E., Morelli G., Russell J. E., Urwin R., Zhang Q., Zhou J., Zurth K., Caugant D. A., Feavers I. M., Achtman M., Spratt B. G. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. USA. 1998;95:3140–3145. [PMC free article: PMC19708] [PubMed: 9501229]
- 27.
- Majewski J., Cohan F. M. Adapt globally, act locally: the effect of selective sweeps on bacterial sequence diversity. Genetics. 1999;152:1459–1474. [PMC free article: PMC1460694] [PubMed: 10430576]
- 28.
- Majewski S. I., Goodwin C. S. Restriction endonuclease analysis of the genome of Campylobacter pylori with a rapid extraction method: evidence for considerable genomic variation. J. Infect. Dis. 1988;157:465–471. [PubMed: 2830341]
- 29.
- Maynard Smith J. The detection and measurement of recombination from sequence data. Genetics. 1999;153:1021–1027. [PMC free article: PMC1460795] [PubMed: 10511575]
- 30.
- Maynard Smith J., Smith N. H., O'Rourke M., Spratt B. G. How clonal are bacteria? Proc. Natl. Acad. Sci. USA. 1993;90:4384–4388. [PMC free article: PMC46515] [PubMed: 8506277]
- 31.
- Maynard S. J., Smith N. H. Detecting recombination from gene trees. Mol. Biol. Evol. 1998;15:590–599. [PubMed: 9580989]
- 32.
- Morelli G., Malorny B., Muller K., Seiler A., Wang J. F., Del Valle J., Achtman M. Clonal descent and microevolution of Neisseria meningitidis during 30 years of epidemic spread. Mol. Microbiol. 1997;25:1047–1064. [PubMed: 9350862]
- 33.
- Mukhopadhyay A. K., Kersulyte D., Jeong J. Y., Datta S., Ito Y., Chowdhury A., Chowdhury S., Santra A., Bhattacharya S. K., Azuma T., Nair G. B., Berg D. E. Distinctiveness of genotypes of Helicobacter pylori in Calcutta, India. J. Bacteriol. 2000;182:3219–3227. [PMC free article: PMC94510] [PubMed: 10809703]
- 34.
- Musser J. M., Kroll J. S., Granoff D. M., Moxon E. R., Brodeur B. R., Campos J., Dabernat H., Frederiksen W., Hamel J., Hammond G., Hoiby E. A., Jonsdottir K. E., Kabeer M., Kallings I., Khan W. N., Kilian M., Knowles K., Koornhof H. J., Law B., Li I., Montgomery J., Pattison P. E., Piffaretti J.-C., Takala A. K., Thong M. L., Wall R. A., Ward J. I., Selander R. K. Global genetic structure and molecular epidemiology of encapsulated Haemophilus influenzae. Rev. Infect. Dis. 1990;12:75–111. [PubMed: 1967849]
- 35.
- Ng E. K., Thompson S. A., Perez-Perez G. I., Kansau I., van der Ende A., Labigne A., Sung J. J., Chung S. C., Blaser M. J. Helicobacter pylori heat shock protein A: serologic responses and genetic diversity. Clin. Diagn. Lab. Immunol. 1999;6:377–382. [PMC free article: PMC103726] [PubMed: 10225839]
- 36.
- Nwokolo C. U., Bickley J., Attard A. R., Owen R. J., Costas M., Fraser I. A. Evidence of clonal variants of Helicobacter pylori in three generations of a duodenal ulcer disease family. Gut. 1992;33:1323–1327. [PMC free article: PMC1379597] [PubMed: 1446853]
- 37.
- Ochman H., Wilson A. C. Evolution in bacteria: evidence for a universal substitution rate in cellular genomes. J. Mol. Evol. 1987;26:74–86. [PubMed: 3125340]
- 38.
- Odenbreit S., Püls J., Sedlmaier B., Gerland E., Fischer W., Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–1500. [PubMed: 10688800]
- 39.
- Owen R. J., Bickley J., Lastovica A., Dunn J. P., Borman P., Hunton C. Ribosomal RNA gene patterns of Helicobacter pylori from surgical patients with healed and recurrent peptic ulcers. Epidemiol. Infect. 1992;108:39–50. [PMC free article: PMC2272182] [PubMed: 1372263]
- 40.
- Pagliaccia C., De Bernard M., Lupetti P., Ji X., Burroni D., Cover T. L., Papini E., Rappuoli R., Telford J. L., Reyrat J. M. The m2 form of the Helicobacter pylori cytotoxin has cell type-specific vacuolating activity. Proc. Natl. Acad. Sci. USA. 1998;95:10212–10217. [PMC free article: PMC21487] [PubMed: 9707626]
- 41.
- Römling U., Schmidt K. D., Tümmler B. Large chromosomal inversions occur in Pseudomonas aeruginosa clone C strains isolated from cystic fibrosis patients. FEMS Microbiol. Lett. 1997;150:149–156. [PubMed: 9163919]
- 42.
- Salama S. M., Jiang Q., Chang N., Sherbaniuk R. W., Taylor D. E. Characterization of chromosomal DNA profiles from Helicobacter pylori strains isolated from sequential gastric biopsy specimens. J. Clin. Microbiol. 1995;33:2496–2497. [PMC free article: PMC228454] [PubMed: 7494058]
- 43.
- Salaun L., Audibert C., Le Lay G., Burucoa C., Fauchere J. L., Picard B. Panmictic structure of Helicobacter pylori demonstrated by the comparative study of six genetic markers. FEMS Microbiol. Lett. 1998;161:231–239. [PubMed: 9570115]
- 44.
- Sawyer S. Statistical tests for detecting gene conversion. Mol. Biol. Evol. 1989;6:526–538. [PubMed: 2677599]
- 45.
- Segal E. D., Cha J., Lo J., Falkow S., Tompkins L. S. Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc. Natl. Acad. Sci. USA. 1999;96:14559–14564. [PMC free article: PMC24475] [PubMed: 10588744]
- 46.
- Selander, R. K., J. Li, and K. Nelson. 1996. Evolutionary genetics of Salmonella enterica, p. 2691–2707. In F. C. Neidhardt (ed.), Escherichi coli and Salmonella: Cellular and Molecular Biology, 2nd ed. ASM Press, Washington, D. C.
- 47.
- Sreevatsan S., Pan X., Stockbauer K. E., Connell N. D., Kreiswirth B. N., Whittam T. S., Musser J. M. Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc. Natl. Acad. Sci. USA. 1997;94:9869–9874. [PMC free article: PMC23284] [PubMed: 9275218]
- 48.
- Stephens J. C. Statistical methods of DNA sequence analysis: detection of intragenic recombination or gene conversion. Mol. Biol. Evol. 1985;2:539–556. [PubMed: 3870876]
- 49.
- Suerbaum S., Maynard Smith J., Bapumia K., Morelli G., Smith N. H., Kunstmann E., Dyrek I., Achtman M. Free recombination within Helicobacter pylori. Proc. Natl. Acad. Sci. USA. 1998;95:12619–12624. [PMC free article: PMC22880] [PubMed: 9770535]
- 50.
- Suerbaum S., Thiberge J.-M., Kansau I., Ferrero R. L., Labigne A. Helicobacter pylori hspA-hspB heat shock gene cluster: nucleotide sequence, expression, putative function and immunogenicity. Mol. Microbiol. 1994;14:959–974. [PubMed: 7715457]
- 51.
- van der Ende A., Pan Z. J., Bart A., van der Hulst R. W., Feller M., Xiao S. D., Tytgat G. N., Dankert J. cagA-positive Helicobacter pylori populations in China and the Netherlands are distinct. Infect. Immun. 1998;66:1822–1826. [PMC free article: PMC108130] [PubMed: 9573056]
- 52.
- van Doorn L. J., Figueiredo C., Megraud F., Pena S., Midolo P., Queiroz D. M., Carneiro F., Vanderborght B., Pegado M. D., Sanna R., de Boer W., Schneeberger P. M., Correa P., Ng E. K., Atherton J., Blaser M. J., Quint W. G. Geographic distribution of vacA allelic types of Helicobacter pylori. Gastroenterology. 1999;116:823–830. [PubMed: 10092304]
- 53.
- van Doorn L. J., Figueiredo C., Sanna R., Blaser M. J., Quint W. G. Distinct variants of Helicobacter pylori cagA are associated with vacA subtypes. J. Clin. Microbiol. 1999;37:2306–2311. [PMC free article: PMC85143] [PubMed: 10364602]
- 54.
- van Doorn L. J., Figueiredo C., Sanna R., Pena S., Midolo P., Ng E. K., Atherton J. C., Blaser M. J., Quint W. G. Expanding allelic diversity of Helicobacter pylori vacA. J. Clin. Microbiol. 1998;36:2597–2603. [PMC free article: PMC105169] [PubMed: 9705399]
- 55.
- Vincent P., Gottrand F., Pernes P., Husson M. O., Lecomte Houcke M., Turck D., Leclerc H. High prevalence of Helicobacter pylori infection in cohabiting children. Epidemiology of a cluster, with special emphasis on molecular typing. Gut. 1994;35:313–316. [PMC free article: PMC1374581] [PubMed: 8150338]
- 56.
- Wang G., Humayun M. Z., Taylor D. E. Mutation as an origin of genetic variability in Helicobacter pylori. Trends Microbiol. 1999;7:488–493. [PubMed: 10603484]
- 57.
- Wang J. T., Sheu J. C., Lin J. T., Wang T. H., Wu M. S. Direct DNA amplification and restriction pattern analysis of Helicobacter pylori in patients with duodenal ulcer and their families. J. Infect. Dis. 1993;168:1544–1548. [PubMed: 8245543]
- 58.
- Whittam T. S., Reid S. D., Selander R. K. Mutators and long-term molecular evolution of pathogenic Escherichia coli O157:H7. Emerg. Infect. Dis. 1998;4:615–617. [PMC free article: PMC2640239] [PubMed: 9866737]
- Helicobacter pylori genome variability in a framework of familial transmission.[BMC Microbiol. 2007]Helicobacter pylori genome variability in a framework of familial transmission.Kivi M, Rodin S, Kupershmidt I, Lundin A, Tindberg Y, Granström M, Engstrand L. BMC Microbiol. 2007 Jun 11; 7:54. Epub 2007 Jun 11.
- Type I restriction-modification loci reveal high allelic diversity in clinical Helicobacter pylori isolates.[Helicobacter. 2010]Type I restriction-modification loci reveal high allelic diversity in clinical Helicobacter pylori isolates.Andres S, Skoglund A, Nilsson C, Krabbe M, Björkholm B, Engstrand L. Helicobacter. 2010 Apr; 15(2):114-25.
- Helicobacter pylori genetic diversity within the gastric niche of a single human host.[Proc Natl Acad Sci U S A. 2001]Helicobacter pylori genetic diversity within the gastric niche of a single human host.Israel DA, Salama N, Krishna U, Rieger UM, Atherton JC, Falkow S, Peek RM Jr. Proc Natl Acad Sci U S A. 2001 Dec 4; 98(25):14625-30. Epub 2001 Nov 27.
- Review [Diversity in genome and epigenome of Helicobacter pylori].[Nihon Saikingaku Zasshi. 2015]Review [Diversity in genome and epigenome of Helicobacter pylori].Furuta Y. Nihon Saikingaku Zasshi. 2015; 70(4):383-9.
- Review Mutation and recombination in Helicobacter pylori: mechanisms and role in generating strain diversity.[Int J Med Microbiol. 2005]Review Mutation and recombination in Helicobacter pylori: mechanisms and role in generating strain diversity.Kraft C, Suerbaum S. Int J Med Microbiol. 2005 Sep; 295(5):299-305.
- Population Genetics - Helicobacter pyloriPopulation Genetics - Helicobacter pylori
Your browsing activity is empty.
Activity recording is turned off.
See more...