

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

The immune system functions by maintaining a delicate balance between the activities of pro-inflammatory and anti-inflammatory pathways. Unbalanced activation of these pathways often leads to the development of serious inflammatory diseases. TNF (Tumor Necrosis Factor) is a key pro-inflammatory cytokine, which can cause several inflammatory diseases when inappropriately up-regulated. Inhibition of TNF activities by using modulatory recombinant proteins has become a successful therapeutic approach to control TNF activity levels but these anti-TNF reagents also have risks and certain limitations. Biological molecules with a different mode of action in regulating TNF biology might provide a clinically useful alternative to the current therapeutics or in some cases might be efficacious in combination with existing anti-TNF therapies. TNF is also a powerful host defense cytokine commonly induced in the host response against various invading pathogens. Many viral pathogens can block TNF function by encoding modulators of TNF, its receptors or downstream signaling pathways. Here, we review the known virus-encoded TNF inhibitors and evaluate their potential as alternative future anti-TNF therapies.
Introduction
Dynamic interactions are set into motion between the host and pathogens whenever they encounter each other. All successful pathogens, including viruses, bacteria and intracellular parasites have adapted diverse mechanisms to counteract the innate and adaptive responses mounted by the host. During this process many have evolved to express specific pathogen-encoded molecules that have regulatory roles in controlling the immune system of the infected host.1-3 These pathogen-derived molecules have often been well-honed by evolutionary selection pressures and can be attractive platform candidates as novel therapeutics to regulate the host immune system in diseases where exacerbated immune or inflammatory cascades have become pathologic to the host.4,5
Pro-inflammatory cytokines like TNFα (here called TNF) play very important roles in orchestrating host defense against invading pathogens, but uncontrolled expression of these cytokines sometimes creates inflammatory diseases in humans if not properly regulated. Various anti-TNF therapeutics, such as neutralizing monoclonal antibodies or Fc fusions of TNF receptor ectodomains, have now entered into the arena of clinical usage to control inappropriate and excessive elaboration of TNF. Virus-encoded TNF inhibitors or modulators of TNF function can be exceedingly potent inhibitors of TNF pro-inflammatory activities.6 Here we discuss whether any of these virus-derived inhibitors might have potential clinical utility as an alternative strategy to dampen TNF-mediated pathologies.
TNF and TNF-Mediated Signaling
TNF is first expressed as a membrane-bound ligand that can be cleaved and secreted as a nonglycosylated trimer of a 17-kDa protein. TNF is predominantly expressed from macrophages, monocytes, CD4+ and CD8+ T-cells, smooth muscle cells, activated NK cells, neutrophils and fibroblasts. TNF production is inducible by a number of diverse stimuli, such as interferons, IL-2 and IL-18. The initial precursor protein, 26 kDa pro-TNF, is translated, translocated to the endoplasmic reticulum, transported to the cell surface via the Golgi apparatus and is then presented on the cell membrane as a homo-trimeric complex. This cell-surface form of TNF can interact with TNF receptors of neighboring cells or it can be cleaved and released from the cell surface as a soluble trimeric ligand by the TNF converting enzyme (TACE). Cleavage and release from the cell surface appears to have some role for the biological properties of the TNF molecule in vivo, but both forms of the ligand can induce potent signaling activities following interaction with the two known TNF receptors in cell culture. For both the cell associated and secreted forms of TNF, ligand trimerization is required for biological activity. Either the cell bound or soluble TNF ligand binds to two structurally distinct receptors: Type I (TNFR1/p55) and Type II (TNFR2/p75), which are present on the membrane of all cell types except erythrocytes. The two receptors differ significantly in their binding affinities with TNF and other TNF-superfamily members, as well as differing in their intracellular signaling pathways. Both receptors have multiple cytoplasmic domains that control their signaling properties but TNFR1 also has an additional intracellular death domain (DD) for its diverse signaling events.7,8
The trimeric TNF ligand binds to the extracellular domain of the receptors, via domains referred to as Cysteine-Rich Domains (CRDs), which induces conformational changes in the receptor and activates the intracellular signaling pathway, which itself can vary according to the cell type. Binding of TNF with TNFR1 leads to the release of the inhibitory protein silencer of death domains (SODD) from TNFR1 intracellular DD.9 Release of SODD allows binding of TRADD (TNFR1-associated death domain protein) to the DD, which can further activate either the apoptotic pathway, via the Fas-associated death domain (FADD) protein, or the pro-inflammatory pathway, via TNF receptor-associated factor 2 (TRAF2) and receptor-interacting protein (RIP), resulting in the activation of nuclear factor-κB (NF-κB) (Fig. 1). In contrast to TNF-R1, TNF-R2 is unable to activate the TRADD/FADD pathway and signals only through the TRAF2-associated pathway. Some studies have indicated the presence of cross-talk between the two receptors, which is likely to be responsible for the net response of a cell upon TNF stimulation.7 It is also possible that other cellular receptors can form complexes with TNF-receptors and thus add yet more levels of complexity in TNF-induced signaling.
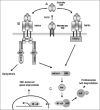
PLAD Domain of TNFRs
The TNFR superfamily members are all Type I transmembrane proteins characterized by the presence of one to six hallmark CRDs. Many members of the TNFR superfamily (e.g., FAS, TNFR1 and TNFR2) exist as pre-assembled oligomers on the cell surface. This preligand assembly of TNFR oligomers is mediated by the preligand assembly domain (PLAD), which resides within the N-terminal cysteine-rich domain of the receptors and is not directly involved in ligand binding.10 PLAD-mediated preligand assembly has also been reported for TRAIL receptors and viral TNFR homologues.11,12 The PLAD domain of TNFR1 is critical in TNF responses, because mutation in the PLAD region reduces NF-κB activation and results in the TNFR-associated periodic syndrome, an autoinflammatory syndrome in man.13 Also, mutation in the PLAD region of FAS has been found to participate in pathogenesis of autoimmune lymphoproliferative syndrome (ALPS), a human genetic disease involving defective apoptosis, lymphocyte accumulation and autoimmunity.14 The mutant form of PLAD appears to inhibit the pre-assembly of FAS chain, thereby blocking the FAS intracellular signaling pathway.14 Recent evidence indicates that PLAD-mediated receptor association regulates cellular responses to TNF-like cytokines, especially in cells of the immune system such as CD4+ and CD8+ T-cells.15 Thus, targeting preligand assembly itself may offer new possibilities for therapeutic intervention in different pathological conditions involving hyperactive TNF signaling. PLAD domain proteins can also effectively prevent TNFR signaling and potently inhibit inflammatory arthritis.16
TNF-Mediated Diseases
The immune system constantly maintains a delicate balance between the pro-inflammatory and anti-inflammatory mediators or cytokines. During many disease states, this balance is lost and the pro-inflammatory cytokines like TNF can become inappropriately upregulated. This, in turn, ultimately induces excessive levels of adhesion molecules on the endothelium, stimulates fibroblast proliferation and recruits leukocytes from the circulation into tissues or sites where they can be pathologic, such as the synovial fluid.17 Enhanced levels of TNF are associated with the development of a variety of inflammatory conditions, like Rheumatoid Arthritis (RA), juvenile RA, Crohn's disease (CD), Ankylosing Spondylitis (AS), Psoriatic Arthritis (PsA), Inflammatory Bowel disease (IBD) and asthma. TNF also can play an indirect role in other inflammatory conditions, as reported in the case of ocular inflammatory development.18 The recent findings that hyperactive systemic inflammation contributes to the development of atherosclerosis and Type 2 diabetes mellitus constitutes a major breakthrough in understanding the mechanisms underlying these conditions. Thus, TNF has been shown to play a key role in many human inflammatory disorders and is considered to be a prime therapeutic target for drug development.
Apart from its direct pro-inflammatory role, TNF also stimulates production of other pro-inflammatory cytokines (such as IL-1 and IL-6), chemokines, reactive oxygen intermediates, nitric oxide and prostaglandins and increases the rate of tissue remodeling by matrix-degrading proteases.19 TNF promotes angiogenesis and osteoclast differentiation and activates osteoclasts that stimulate bone lysis, leading to joint erosions particularly at the marginal surfaces.20 TNF also increases the rate of tissue remodeling by matrix-degrading proteases and directly mediates pain, fever and cachexia. Additionally, TNF has a significant role in lipid metabolism. In animal models, administration of exogenous TNF leads to severe impairment of glucose tolerance and insulin sensitivity.21 Thus, TNF might serve as a therapeutic target in these disorders as well.
Current Anti-TNF Therapies in Humans
Several recombinant protein-based inhibitors of TNF activity have been developed for clinical therapy. Three of these, Etanercept (Enbrel), infliximab (Remicade) and adalimumab (Humira) have already been approved for clinical use in various inflammatory diseases. Etanercept is a dimeric fusion protein consisting of two extracellular domains of the human TNFR2, linked to the Fc portion of a Type 1 human immunoglobulin (IgG1). The Fc portion helps to maintain the molecule in the circulation22 and it has a relatively short half life of 4-5 days. Infliximab is a chimeric mAb, composed of human constant regions of IgG1, with murine variable regions. It binds to both soluble and membrane TNF with high affinity and exhibits lower nonspecific effects on other pathways.23 Infliximab has a circulating half life of 8-10 days. Adalimumab is a complete human IgG1 anti-TNF mAb that binds to both soluble and membrane bound TNF with high affinity. It has a relatively longer half life of 12-14 days. Several other protein based anti-TNF therapeutics are also currently in clinical trials. Numerous studies have been conducted to understand the mechanism of action of the various anti-TNF therapeutics in immune-mediated diseases. In addition to neutralization of soluble TNF, these drugs clearly affect diverse intracellular signaling which regulate cell cycle arrest, apoptosis, or suppression of cytokine production.24
Although the current protein-based TNF inhibitors have demonstrated ligand-inhibitory efficacy, they can also exhibit potentially serious adverse effects such as a greater predisposition towards secondary infections, congestive heart failure, neurologic changes (demyelination), lymphomas, re-exacerbation of latent tuberculosis and problems related to autoimmunity such as lupus-like syndrome.25 With infliximab, acute allergic reactions are seen in approximately 5% of intravenous infusions as well.
Safety Issues with Current Anti-TNF Therapies
Some of the major safety considerations regarding the long term use of TNF antagonists include infections, autoimmune disease, demyelinating disease, malignancies and congestive heart failure. Because TNF has significant role in host defense to both bacterial and viral invasion, one of the main concerns with anti-TNF therapy is risk of infection. Nevertheless, patients with RA who received anti-TNF agents do not show a significant increase in the overall rate of infections during treatment but there are case reports of pulmonary listeriosis,26 pulmonary aspergillosis,27 Pneumocystis carinii pneumonia28 and reactivated histoplasmosis29 in some patients. Tuberculosis (TB) recrudescence has been the most common serious infection observed in patients receiving TNF antagonists.30 However, the incidence of TB is also influenced by age, concomitant immunosuppressive regimens, socioeconomic status and geography.31 Screening of patients for the possibility of latent TB and other related pathogens before treatment would further reduce the rate of infections exacerbated by treatment with TNF inhibitors.
The regulatory role of TNF with antigen-presenting cell function may be associated with autoimmune diseases arising in some patients treated with TNF antagonists. Anti-TNF treatment very often results in the development of antinuclear antibodies and anti- double-stranded DNA antibodies.32 Some of the reported autoimmune diseases associated with anti-TNF therapy are systemic lupus erythematosus (SLE) and different forms of vasculitis.33 Among the neurological diseases reported, demyelinating disease is most common in patients with inflammatory arthritis. Congestive heart failure has been reported, mostly in RA patients. Although not common, anti-TNF drugs may also increase incidence of solid tumor development in patients with RA.33 In many cases the complications from anti-TNF treatment depends on the specific biological agent used.
Recently and for the first time, it has been reported that perforating folliculitis (a type of perforating dermatosis characterized by trans-epithelial elimination of dermal structures) is associated with the administration of the TNF inhibitors, infliximab and etanercept, in a patient suffering from RA and pulmonary fibrosis.34 The possible reason is that TNF directly inhibits fibronectin production and promotes its degradation through stimulation of several metalloproteinases.35 Therefore, the blockade of TNF may induce fibronectin accumulation, favoring in some way the perforating phenomenonobserved in these patients.
Viral TNF Inhibitors as Alternative Therapeutics
Viruses have adapted diverse strategies to neutralize TNF and TNF-mediated responses by targeting almost every step of the TNF response pathway.6,36,37 The virus-encoded modulators can either directly bind the ligand and/or receptor, or components of the TNFR signaling pathway, to inhibit the TNF response (Fig. 1). Some viral regulators have adapted a strategy of down-regulating the expression of cell surface TNFRs. Components of the TNF signal transduction pathway are also a recurrent target for viral immune evasion because of their central roles in mounting innate and adaptive immune responses.36,37 Some of the known viral immune modulating molecules directly bind and inhibit the function of TNF ligand (Table 1) whereas others modulate downstream TNFR functions (Table 2).
Table 1
Poxvirus-encoded TNF inhibitors.
Table 2
Virus-encoded TNF modulators.
So far, poxviruses are the only viruses that express viral modulators that can bind and sequester extracellular TNF prior to its TNFR engagement.6 Two distinct classes of extracellular poxvirus TNF-binding proteins have been identified: those that resemble the mammalian TNFRs, termed vTNFRs and a recently characterized family of proteins that resemble the mammalian MHC class I heavy chain, termed vTNF-BPs, that bind and inhibit TNF with unusually tight affinity.38,39
Viral TNFR Homologues
Among these vTNFRs are the T2-like family members encoded by Leporipoxviruses and the cytokine response modifier (Crm) family members encoded by Orthopoxviruses.40,41 Like their mammalian counterparts, poxvirus vTNFR superfamily proteins comprise a tandem array of CRDs. These CRDs are each composed of two distinct structural modules that can adopt one of a number of conserved conformations (primarily A or B), can contain zero to two disulphide bonds and are named according to topology and disulphide bond count (A1, A2, etc.). Individual poxviruses can express different numbers of TNFR-like molecules: cowpox virus (CPXV) expresses up to five TNFR superfamily molecules, cytokine response modifier B (CrmB), CrmC, CrmD, CrmE and vCD30,42-46 while variola virus (VARV) and monkeypox virus (MPXV), which causes a smallpox-like disease in humans, express only one, CrmB,47,48 as does Ectromelia virus, a mouse pathogen that cause mousepox, which expresses CrmD only.49 These CrmB, C, D, E proteins bear no structural similarity to CrmA which is a cross-class serpin. Poxviral TNFR superfamily members differ from their mammalian counterparts in that they lack a transmembrane region and cytoplasmic domain for signaling and are transported to the extracellular millieu. Vaccinia virus (VACV) strains USSR, Lister and Evans encode two functional vTNFR superfamily members: CrmC and CrmE.50 These are the smallest of the known poxvirus vTNFRs, containing an N-terminal signal peptide for secretion into the extracellular matrix, three CRDs and a short C-terminal extension.
In some cases, poxvirus-encoded vTNFR homologues have additional properties in addition to the binding and inhibiting of TNF functions. This has been demonstrated in case of myxoma virus encoded M-T2 protein, which has a second anti-apoptotic role.12 In case of CrmE, it has recently been shown that the protein also possesses chemokine binding properties.47 Thus, unlike the engineered commercial TNF inhibitors currently used in humans, the virus-encoded modulators sometime possess additional anti-inflammatory properties above and beyond just TNF inhibition.
CrmE
All members of the poxvirus-encoded vTNFR superfamily of proteins lack a C-terminal transmembrane domain like the ones in TNFR1 or TNFR2. CrmE from CPXV is expressed as a secreted 18kDa protein. CrmE binds to human, mouse and rat TNF but not to any other member of TNF superfamily.45 Biologically, it protects cells from the cytolytic activity of human TNF by binding to it with high affinity. The CrmE ortholog from vaccinia virus has also the same properties as CPXV ortholog.51 Cells infected with VACV strains that encode CrmE (e.g., strain Lister) display both soluble and cell surface associated CrmE activity.51 VACV CrmE inhibited the cytotoxic and apoptotic activity of only human TNF in vitro. The expression of CrmE by a VACV strain (VACV WR) that does not normally encode a viral TNFR enhances virus virulence.51 It was predicted that the CrmE members structurally resemble TNFR2. The structure of CrmE protein from VACV strain Lister has been solved recently52 which shares significant sequence similarity with mammalian Type 2 TNF receptors (TNFSFR1B, p75; TNFR2). The bacterially expressed and purified CrmE is a monomer in solution and forms a stable complex with recombinant human TNF to form a heterohexamer. The structure confirms that CrmE adopts the canonical TNFR fold but only one of the two “ligand binding” loops of TNFR1 is conserved in CrmE, suggesting a mechanism for the higher affinity of poxvirus vTNFRs for TNF over LT-α.52
CrmB and CrmD
The CrmB from VARV, MPXV and CPXV differ in their efficiencies of inhibition of cytotoxic effects of human, mouse or rabbit TNF.48 The CrmB and CrmD, in addition to their CRD, have an extended C terminus. The TNF binding activity is exclusively located in the N-terminal CRDs of these proteins. Surface Plasmon Resonance (SPR) screening of CrmB with different cytokines and chemokines identified that it also binds to human chemokines. The chemokine binding region is exclusively located to the C-terminal CTD. This suggests that CrmB and CrmD have distinct and independent binding sites for TNF and chemokines and might thus have a broader role as an anti-inflammatory modulator. The CTD of the variola version has been named the SECRET (smallpox virus-encoded chemokine receptor) domain.47 The CrmB proteins from VARV, MPXV and CPXV were tested for their ability to protect BALB/c mice against LPS induced endotoxic shock. VARV-CrmB protein exhibited a more efficient protective effect with an increase in the animal survival rate. This protection could be the combined effect of TNF-binding and chemokine binding activity of the N-terminal CRDs and C-terminal SECRET domain respectively.48 However, further in vivo studies are required to understand the pharmacologic properties of these Crm proteins.
Viral PLAD Like Domain
The role of PLAD domain in regulating TNF-like cytokine signaling has been recently studied in the viral TNFR homologue from myxoma virus. The myxoma virus encoded protein T2 (M-T2) binds and inhibits only rabbit TNF.53 Rabbits infected with M-T2 knockout myxoma virus exhibited a markedly attenuated disease progression compared to rabbits infected with the wild type myxoma virus, suggesting that M-T2 is a viral virulence factor. Another function of M-T2 is to prevent apoptosis of myxoma virus infected rabbit CD4+ RL5 T-cells.54 RL5 cells infected with T2 KO virus undergo apoptosis and prevent optimal virus replication as compared to WT myxoma virus infected cells. Deletion analysis of M-T2 demonstrated that the intracellular part of the protein possesses anti-apoptotic activity because active purified M-T2 protein added to the culture supernatants of vMyxT2ko-infected RL5 cells failed to rescue these cells from virus-induced apoptosis.55 Thus, M-T2 has dual roles in two different forms; secreted M-T2 binds and inhibits rabbit TNF, whereas a different N-terminal domain of the intracellular M-T2 protein blocks virus-infected lymphocyte apoptosis.41
The anti-apoptotic role of M-T2 as inhibitor of TNFR-mediated cell death has been further investigated recently. The first evidence came from the observation that human Jurkat T-cells expressing M-T2 were resistant to TNF and TNFR-induced cell death.12 Note that intact M-T2 protein neither binds nor inhibits human TNF. This M-T2-mediated inhibition of TNFR-induced cell death requires the PLAD domain located in the N-terminus of the M-T2 protein. Further, biochemical and colocalization studies using fluorescently tagged receptors demonstrated that M-T2 interacts with human TNFR1 and TNFR2 via the PLAD.12 Thus, the M-T2 CRD domain interacts with TNF in a species-dependent fashion whereas the M-T2 PLAD domain binds both TNFRs in a species-independent manner. A version of the PLAD domain is present and conserved in all poxvirus T2-like proteins. It is entirely possible that the viral PLAD domain might also interact with other host proteins, in order to mediate its anti-apoptotic role. Viral PLAD domains in theory could be utilized as components of future anti-TNF therapeutics if the issue of protein delivery to the appropriate intracellular location could be addressed.
Viral TNF-Binding Proteins Unrelated to Host TNFRs
Another class of TNF inhibitors has been identified from certain members of poxviruses which do not encode conventional vTNFR orthologs. The viral TNF binding protein (vTNF-BP) was first identified in Tanapox virus (TPV), a member of the Yatapoxvirus.38 Functional orthologs are also present in other member of Yatapoxviruses, Yaba-like disease virus (YLDV) and Yaba monkey tumor virus (YMTV) and a version is also present in swinepox virus.39 Unlike poxvirus vTNFRs, only one copy of these vTNF-BPs inhibitors are present per genome. This class of vTNF-BPs exhibit closest amino acid sequence similarity to MHC class I molecules but, unlike the cellular counterpart, the viral protein lacks a transmembrane domain. The vTNF-BPs tested to date bind to TNFs from different species with diverse affinities. The TPV-encoded vTNF-BP, called TPV-2L, binds to human TNF with very high affinity (Kd, 43pM) which is the highest affinity reported to date among the known protein TNF inhibitors.38 TPV-2L can also bind and inhibit monkey and canine TNF with high affinity. The vTNF-BP encoded by YMTV, called YMTV-2L, can also bind and inhibit human and monkey TNF.39 Binding of TPV-2L and YMTV-2L with TNF also inhibit the TNF-mediated signaling pathway as measured by the degradation of IκBα.39 Interestingly, the orthologous vTNF-BP from Swinepox virus exclusively binds and inhibits only porcine TNF.39 TPV-2L and YMTV-2L have very good potential as anti-TNF agents, in part because their very high affinity for human TNF suggests they could achieve TNF inhibition at pharmacologically lower doses than the currently used TNF inhibitors. Although vTNF-BPs share no sequence relationship with TNFRs, the folding clearly results in very high affinity binding with human TNF. Using human TNF mutants which possess differential binding for TNFR1 and TNFR2, it was demonstrated that the interacting residues for human TNF are different for TNFRs compared to these vTNF-BPs.39 However, solving the crystal structure of these class of TNF inhibitors complexed with TNF would shed more light on their function and future potential as therapeutics.
Viral Proteins That Modulate TNF Receptors and Regulate Downstream Signaling
Viral proteins which modulate the TNF receptor or downstream components of the signaling pathway can also in theory be developed as potential anti-TNF therapeutics. Down-regulation of TNFR on the cell surface is another anti-TNF mechanism adapted by many viral proteins. The adenovirus encoded receptor internalization and degradation (RID) complex associated proteins down-regulate the surface levels of TNFR1 and thereby inhibit NF-κB activation. The adenovirus early transcription region 3 (Ad E3) encodes at least seven proteins, five of which block the acquired or innate immune response. Three of these, Ad E3-14.7K, Ad E3-10.4K and Ad E3-14.5K, impose inhibitory effects on the TNF pathway.56 Two of these proteins, 10.4K (RIDα) and 14.5K (RIDβ), form the hetero-trimeric complex RID in the plasma membrane which inhibits signaling through TNFR1. RID down-regulates surface TNFR1 levels by reducing the assembly of TNFR1 signaling complex and thus inhibiting TNF-induced activation of NF-κB. In terms of the NF-κB pathway, RID blocks the association of members of the IKK complex, as well as the protein kinase RIP, with the TNFR1.56 From the RID complex, RIDβ directly interacts with TNFR1 and its tyrosine sorting motif plays a major role in the down-regulation of TNFR1 by a clathrin-dependent process where TNFR1 is degraded by an endosomal/lysosomal pathway.57 In addition to TNFR1, RID can also down-regulate the expression of FAS,58 TNF-related apoptosis-inducing ligand receptor 1 (TRAIL-R1 and R2)59,60 as well as epidermal growth factor receptor (EGFR)61 in several cell lines.
The down-regulation of TNFR1 by RID also inhibits TNF-induced secretion of chemokines. The anti-inflammatory role of RID has been demonstrated using LPS treatment as the pro-inflammatory stimulus. RID can inhibit LPS-induced signaling pathway without affecting the expression of the LPS receptor, Toll-like receptor 4 (TLR4).62 This suggests that RID also has intracellular targets that inhibit signal transduction and chemokine expression without receptor down-regulation. The potential use of RID as therapeutic immunomodulator has been tested in vivo where RID facilitates transplantation of allogenic pancreatic cells63 and decreases autoimmune Type I diabetes incidence in NOD mice.64,65
Human Papillomaviruses (HPVs), the major cause of cervical cancer, can infect various human epithelial tissues where they inhibit TNF induced apoptosis. HPV16 encodes two oncogene products, E6 and E7, that associate with other cellular proteins involved in cell proliferation and apoptosis and thereby modulate their function. HPV16 E6 protein selectively binds to TNFR1 and affects the transmission of pro-apoptotic signals triggered by TNF.66 E6 binds to the C-terminal 41 amino acids of TNFR1 and inhibits binding of TRADD to TNFR1 and thereby blocks formation of the death-inducing signaling complex (DISC). This inhibition subsequently blocks transmission of apoptotic signals by inhibiting the activation of initiator caspases such as caspase 8. E6-mediated protection against TNF-induced apoptosis occurs in cells of different species (mouse and human) and tissues (fibroblast, osteosarcoma and histiocyte/monocyte). Both E6 and E7 of HPV16 increased the transcription of cIAP1 and cIAP2 by upregulation of NF-κB-expression and confer resistance to TNF in human keratinocytes.67
Cytomegalovirus (CMV) encodes proteins with diverse immunomodulatory functions. Human and murine CMV have developed mechanisms to evade the TNF-induced antiviral state by dysregulating TNFRs. HCMV infection of THP1 cells reduced the level of TNFR1 on the cell surface by accumulating the receptor pool in the trans-Golgi network.68 Time course analysis and drug inhibition studies suggest that viral early gene products may target trafficking of TNFR1.68 MCMV infection blocked TNF-induced nuclear translocation of NF-κB, which decreased the level of both TNFR1 and TNFR2 in bone marrow derived macrophages.69 This was mediated by expression of still-unknown viral immediate early and/or early genes. Identification of the viral molecule(s) responsible could yield a potential therapeutic drug target.
Poliovirus noncapsid protein 3A is a multifunctional viral protein involved in poliovirus RNA replication.70 One of its functions is suppression of protein trafficking between the ER and Golgi apparatus. In infected cells, it affects the intracellular trafficking of TNFR and induces TNF resistance by eliminating TNFRs from the plasma membrane.71 This 3A-protein-mediated inhibition of ER to Golgi traffic of TNFR was limited to poliovirus and coxsackievirus B3. Further investigation is required to understand whether this inhibition is selective for TNFR or not.
The hepatitis C virus (HCV) is the major cause of non-A, nonB hepatitis, which often leads to liver cirrhosis and hepatocellular carcinoma. Two HCV-encoded proteins, nonstructural protein 5A (NS5A) and NS5B, have regulatory roles in TNF signaling and both have potential as anti-TNF therapeutics. Both NS5A and NS5B are essential components of the HCV replication complex. NS5A is a multifunctional phosphoprotein and inhibits TNF-induced signaling using multiple mechanisms. In HepG2 cells expression of NS5A protected these cells from TNF-induced cell death.72 NS5A binds to the TNFR1 signaling complex through its interaction with TRAF2 and subsequently inhibits TRAF2-dependent NF-κB activation, thereby sensitizing the cells to TNF-induced cytotoxicity.73 In another study using NS5A transgenic mice, it has been demonstrated that NS5A interacts with TRADD, inhibiting its association with FADD and TNF-mediated apoptosis, resulting in persistent infection.74 The inhibition of intrinsic apoptotic signals is mediated by the putative BH (Bcl-2 homology) domain of NS5A, which allows it to bind to the pro-apoptotic protein Bax, rendering cells refractory to certain pro-apoptotic agonists.75 The HCV NS5B protein is a membrane-associated phosphoprotein that possesses an RNA dependent RNA polymerase activity.76 NS5B inhibited TNF-induced NF-κB activation in HEK293 and hepatic cells. This inhibition is mediated by its direct interaction with IKKα.
Cell Signaling Inhibitors from Viruses That Inhibit Activation of NF-κB
The ideal candidate for a virus-derived anti-TNF therapeutic would be more specific and less toxic to the human system than current therapies. Several viral inhibitors can specifically block intracellular signaling, often resulting in reduced TNF production or its TNFR-dependant effects. Activation of NF-κB is an early event that occurs within minutes after exposure to TNF and plays important role in inflammation, regulation of cell proliferation, activation and survival.77 This activation process has turned out to be an attractive target for viruses to escape immune defenses and many viruses have evolved specific gene products to inhibit the TNF-induced NF-κB activation.
Vaccinia virus encoded protein N1L, a viral virulence factor, inhibits signaling through NF-κB from both TNF and LTα. This N1L-mediated inhibition of NF-κB occurs by association with IKK-γ and inhibition of IKK-α and IKK-β.78 N1L also inhibits IRF3 signaling and thus might play a broad role as viral immunomodulator of innate immunity. However, N1L might have even additional biologic roles. N1L inhibits apoptosis and interacts with pro-apoptotic Bcl2 proteins like Bid, Bax, Bad, Bim and Bak.79,80 Recent crystal structure data of N1L suggest that it belongs to Bcl2 family of anti-apoptotic proteins, although no amino acid sequence similarity to these cellular proteins was observed by sequence analysis.80 The structure also revealed that N1L contains a surface groove that resembles the BH3-binding grooves of other Bcl-2 proteins. In another report, it was shown that N1L also inhibits the release of pro-inflammatory cytokines like TNF, IL-1β, IFNα and IFNβ and the anti-inflammatory cytokine IL-10 from human primary monocytes.81
Another vaccinia virus-encoded protein, K1L, inhibits NF-κB activation and could be a potential inhibitor of TNF-mediated responses. K1L inhibits NF-κB by preventing IκBα degradation, probably by interfering directly with IKK to prevent phosphorylation or indirectly by hampering kinases that act upstream of IKK.82 K1L was originally identified as one of the vaccinia host range genes required for viral replication in certain human cells and rabbit kidney cells RK13. The host range function of K1L is associated with the ankyrin repeats present in this protein.83
African swine fever virus (ASFV) A238L, which is an ankyrin-repeat–containing homolog of host IκB (ASFV-IκB), binds to NF-κB following degradation of host IκB and inhibits the nuclear transportation of NF-κB.84 It also inhibits TNF-induced gene expression. The mechanism of regulation of TNF lies in the promoter region that involves CREB binding protein (CBP)/p300 function. Localization of A238L in the nucleus and binds to the cyclic AMP-responsive element and displace the CBP/p300 coactivators.85
Conclusions
Given the sophistication with which viral proteins are currently known to exercise their anti-TNF properties, it would be negligent to ignore the lessons they can impart about how to inhibit TNF and functionally disable pro-inflammatory cascades. Current biotechnology has generated efficient protein-based TNF inhibitors with Kd values in the nanomolar range, but viral TNF-BPs can bind and inhibit human TNF with more than an order of magnitude higher affinity than any commercial TNF inhibitor. At this point, it is unknown if any specific vTNF inhibitors will be more or less immunogenic then the clinical TNF blockade reagents currently in use, but experience has shown that immunogenicity issues are not resolvable with animal models alone. Realistically, the potential of vTNF inhibitors for treating inflammatory disorders will only be settled with properly designed clinical trials.
Acknowledgements
GM is supported by an International Scholarship from HHMI and AL holds the Ethel Smith Endowed Chair in Vasculitis. GM and AL are cofounders of Viron Therapeutics, which is developing viral proteins as anti-inflammatory therapeutics.
References
- 1.
- Finlay BB, McFadden G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell. 2006;124(4):767–782. [PubMed: 16497587]
- 2.
- Roy CR, Mocarski ES. Pathogen subversion of cell-intrinsic innate immunity. Nat Immunol. 2007;8(11):1179–1187. [PubMed: 17952043]
- 3.
- Unterholzner L, Bowie AG. The interplay between viruses and innate immune signaling: Recent insights and therapeutic opportunities. Biochem Pharmacol. 2008;75(3):589–602. [PubMed: 17868652]
- 4.
- Lucas A, McFadden G. Secreted immunomodulatory viral proteins as novel biotherapeutics. J Immunol. 2004;173(8):4765–4774. [PubMed: 15470015]
- 5.
- Fallon PG, Alcami A. Pathogen-derived immunomodulatory molecules: future immunotherapeutics? Trends Immunol. 2006;27(10):470–476. [PubMed: 16920025]
- 6.
- Rahman MM, McFadden G. Modulation of tumor necrosis factor by microbial pathogens. PLoS Pathog. 2006;2(2):e4. [PMC free article: PMC1383482] [PubMed: 16518473]
- 7.
- Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3(9):745–756. [PubMed: 12949498]
- 8.
- Hehlgans T, Pfeffer K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology. 2005;115(1):1–20. [PMC free article: PMC1782125] [PubMed: 15819693]
- 9.
- Takada H, Chen NJ, Mirtsos C, et al. Role of SODD in regulation of tumor necrosis factor responses. Mol Cell Biol. 2003;23(11):4026–4033. [PMC free article: PMC155221] [PubMed: 12748303]
- 10.
- Chan FK, Chun HJ, Zheng L, et al. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science. 2000;288(5475):2351–2354. [PubMed: 10875917]
- 11.
- Clancy L, Mruk K, Archer K, et al. Preligand assembly domain-mediated ligand-independent association between TRAIL receptor 4 (TR4) and TR2 regulates TRAIL-induced apoptosis. Proc Natl Acad Sci USA. 2005;102(50):18099–18104. [PMC free article: PMC1312398] [PubMed: 16319225]
- 12.
- Sedger LM, Osvath SR, Xu XM, et al. Poxvirus tumor necrosis factor receptor (TNFR)-like T2 proteins contain a conserved preligand assembly domain that inhibits cellular TNFR1-induced cell death. J Virol. 2006;80(18):9300–9309. [PMC free article: PMC1563942] [PubMed: 16940541]
- 13.
- Siebert S, Fielding CA, Williams BD, et al. Mutation of the extracellular domain of tumour necrosis factor receptor 1 causes reduced NF-kappaB activation due to decreased surface expression. FEBS Lett. 2005;579(23):5193–5198. [PubMed: 16162344]
- 14.
- Siegel RM, Frederiksen JK, Zacharias DA, et al. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science. 2000;288(5475):2354–2357. [PubMed: 10875918]
- 15.
- Chan FK. Three is better than one: preligand receptor assembly in the regulation of TNF receptor signaling. Cytokine. 2007;37(2):101–107. [PMC free article: PMC1965282] [PubMed: 17449269]
- 16.
- Deng GM, Zheng L, Chan FK, et al. Amelioration of inflammatory arthritis by targeting the preligand assembly domain of tumor necrosis factor receptors. Nat Med. 2005;11(10):1066–1072. [PubMed: 16170321]
- 17.
- Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001;344(12):907–916. [PubMed: 11259725]
- 18.
- Theodossiadis PG, Markomichelakis NN, Sfikakis PP. Tumor necrosis factor antagonists: preliminary evidence for an emerging approach in the treatment of ocular inflammation. Retina. 2007;27(4):399–413. [PubMed: 17420690]
- 19.
- Tak PP, Taylor PC, Breedveld FC. et al. Decrease in cellularity and expression of adhesion molecules by anti-tumor necrosis factor alpha monoclonal antibody treatment in patients with rheumatoid arthritis. Arthritis Rheum. 1996;39(7):1077–1081. [PubMed: 8670314]
- 20.
- Goldring SR, Gravallese EM. Pathogenesis of bone lesions in rheumatoid arthritis. Curr Rheumatol Rep. 2002;4(3):226–231. [PubMed: 12010607]
- 21.
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. [PubMed: 7678183]
- 22.
- Mohler KM, Torrance DS, Smith CA, et al. Soluble tumor necrosis factor (TNF) receptors are effective therapeutic agents in lethal endotoxemia and function simultaneously as both TNF carriers and TNF antagonists. J Immunol. 1993;151(3):1548–1561. [PubMed: 8393046]
- 23.
- Harriman G, Harper LK, Schaible TF. Summary of clinical trials in rheumatoid arthritis using infliximab, an anti-TNFalpha treatment. Ann Rheum Dis. 1999;58(Suppl 1):I61–64. [PMC free article: PMC1766581] [PubMed: 10577975]
- 24.
- Wong M, Ziring D, Korin Y, et al. TNF α blockade in human diseases: Mechanisms and future directions. Clin Immunol. 2007;126(2): 121–136. [PMC free article: PMC2291518] [PubMed: 17916444]
- 25.
- Palladino MA, Bahjat FR, Theodorakis EA, et al. Anti-TNF-α therapies: the next generation. Nat Rev Drug Discov. 2003;2(9):736–746. [PubMed: 12951580]
- 26.
- Slifman NR, Gershon SK, Lee JH, et al. Listeria monocytogenes infection as a complication of treatment with tumor necrosis factor alpha-neutralizing agents. Arthritis Rheum. 2003;48(2):319–324. [PubMed: 12571839]
- 27.
- De Rosa FG, Shaz D, Campagna AC, et al. Invasive pulmonary aspergillosis soon after therapy with infliximab, a tumor necrosis factor-alpha-neutralizing antibody: a possible healthcare-associated case? Infect Control Hosp Epidemiol. 2003;24(7):477–482. [PubMed: 12887234]
- 28.
- Tai TL, O'Rourke KP, McWeeney M, et al. Pneumocystis carinii pneumonia following a second infusion of infliximab. Rheumatology (Oxford). 2002;41(8):951–952. [PubMed: 12154220]
- 29.
- Lee JH, Slifman NR, Gershon SK, et al. Life-threatening histoplasmosis complicating immunotherapy with tumor necrosis factor alpha antagonists infliximab and etanercept. Arthritis Rheum. 2002;46(10):2565–2570. [PubMed: 12384912]
- 30.
- Gomez-Reino JJ, Carmona L, Angel Descalzo M. Risk of tuberculosis in patients treated with tumor necrosis factor antagonists due to incomplete prevention of reactivation of latent infection. Arthritis Rheum. 2007;57(5):756–761. [PubMed: 17530674]
- 31.
- Ellerin T, Rubin RH, Weinblatt ME. Infections and anti-tumor necrosis factor alpha therapy. Arthritis Rheum. 2003;48(11):3013–3022. [PubMed: 14613261]
- 32.
- Charles PJ, Smeenk RJ, De Jong J, et al. Assessment of antibodies to double-stranded DNA induced in rheumatoid arthritis patients following treatment with infliximab, a monoclonal antibody to tumor necrosis factor alpha: findings in open-label and randomized placebo-controlled trials. Arthritis Rheum. 2000;43(11):2383–2390. [PubMed: 11083258]
- 33.
- Hyrich KL, Silman AJ, Watson KD, et al. Anti-tumour necrosis factor alpha therapy in rheumatoid arthritis: an update on safety. Ann Rheum Dis. 2004;63(12):1538–1543. [PMC free article: PMC1754871] [PubMed: 15242866]
- 34.
- Gilaberte Y, Coscojuela C, Vazquez C, et al. Perforating folliculitis associated with tumour necrosis factor-alpha inhibitors administered for rheumatoid arthritis. Br J Dermatol. 2007;156(2):368–371. [PubMed: 17223880]
- 35.
- Morgan MB, Truitt CA, Taira J, et al. Fibronectin and the extracellular matrix in the perforating disorders of the skin. Am J Dermatopathol. 1998;20(2):147–154. [PubMed: 9557783]
- 36.
- Benedict CA. Viruses and the TNF-related cytokines, an evolving battle. Cytokine Growth Factor Rev. 2003;14(3-4):349–357. [PubMed: 12787571]
- 37.
- Benedict CA, Banks TA, Ware CF. Death and survival: viral regulation of TNF signaling pathways. Curr Opin Immunol. 2003;15(1):59–65. [PubMed: 12495734]
- 38.
- Brunetti CR, Paulose-Murphy M, Singh R, et al. A secreted high-affinity inhibitor of human TNF from Tanapox virus. Proc Natl Acad Sci USA. 2003;100(8):4831–4836. [PMC free article: PMC153641] [PubMed: 12676996]
- 39.
- Rahman MM, Barrett JW, Brouckaert P, et al. Variation in ligand binding specificities of a novel class of poxvirus-encoded tumor necrosis factor-binding protein. J Biol Chem. 2006;281(32):22517–22526. [PubMed: 16782702]
- 40.
- Cunnion KM. Tumor necrosis factor receptors encoded by poxviruses. Mol Genet Metab. 1999;67(4):278–282. [PubMed: 10444338]
- 41.
- Xu X, Nash P, McFadden G. Myxoma virus expresses a TNF receptor homolog with two distinct functions. Virus Genes. 2000;21(1-2):97–109. [PubMed: 11022793]
- 42.
- Hu FQ, Smith CA, Pickup DJ. Cowpox virus contains two copies of an early gene encoding a soluble secreted form of the Type II TNF receptor. Virology. 1994;204(1):343–356. [PubMed: 8091665]
- 43.
- Smith CA, Hu FQ, Smith TD, et al. Cowpox virus genome encodes a second soluble homologue of cellular TNF receptors, distinct from CrmB, that binds TNF but not LT alpha. Virology. 1996;223(1):132–147. [PubMed: 8806547]
- 44.
- Loparev VN, Parsons JM, Knight JC, et al. A third distinct tumor necrosis factor receptor of orthopoxviruses. Proc Natl Acad Sci USA. 1998;95(7):3786–3791. [PMC free article: PMC19915] [PubMed: 9520445]
- 45.
- Saraiva M, Alcami A. CrmE, a novel soluble tumor necrosis factor receptor encoded by poxviruses. J Virol. 2001;75(1):226–233. [PMC free article: PMC113916] [PubMed: 11119592]
- 46.
- Panus JF, Smith CA, Ray CA, et al. Cowpox virus encodes a fifth member of the tumor necrosis factor receptor family: a soluble, secreted CD30 homologue. Proc Natl Acad Sci USA. 2002;99(12):8348–8353. [PMC free article: PMC123070] [PubMed: 12034885]
- 47.
- Alejo A, Ruiz-Arguello MB, Ho Y, et al. A chemokine-binding domain in the tumor necrosis factor receptor from variola (smallpox) virus. Proc Natl Acad Sci USA. 2006;103(15):5995–6000. [PMC free article: PMC1458686] [PubMed: 16581912]
- 48.
- Gileva IP, Nepomnyashchikh TS, Antonets DV, et al. Properties of the recombinant TNF-binding proteins from variola, monkeypox and cowpox viruses are different. Biochim Biophys Acta. 2006;1764(11):1710–1718. [PMC free article: PMC9628946] [PubMed: 17070121]
- 49.
- Smith VP, Alcami A. Expression of secreted cytokine and chemokine inhibitors by ectromelia virus. J Virol. 2000;74(18):8460–8471. [PMC free article: PMC116357] [PubMed: 10954546]
- 50.
- Alcami A, Khanna A, Paul NL, et al. Vaccinia virus strains Lister, USSR and Evans express soluble and cell-surface tumour necrosis factor receptors. J Gen Virol. 1999;80(Pt 4):949–959. [PubMed: 10211965]
- 51.
- Reading PC, Khanna A, Smith GL. Vaccinia virus CrmE encodes a soluble and cell surface tumor necrosis factor receptor that contributes to virus virulence. Virology. 2002;292(2):285–298. [PubMed: 11878931]
- 52.
- Graham SC, Bahar MW, Abrescia NG, et al. Structure of CrmE, a virus-encoded tumour necrosis factor receptor. J Mol Biol. 2007;372(3):660–671. [PubMed: 17681535]
- 53.
- Schreiber M, McFadden G. The myxoma virus TNF-receptor homologue (T2) inhibits tumor necrosis factor-alpha in a species-specific fashion. Virology. 1994;204(2):692–705. [PubMed: 7941338]
- 54.
- Macen JL, Graham KA, Lee SF, et al. Expression of the myxoma virus tumor necrosis factor receptor homologue and M11L genes is required to prevent virus-induced apoptosis in infected rabbit T-lymphocytes. Virology. 1996;218(1):232–237. [PubMed: 8615027]
- 55.
- Schreiber M, Sedger L, McFadden G. Distinct domains of M-T2, the myxoma virus tumor necrosis factor (TNF) receptor homolog, mediate extracellular TNF binding and intracellular apoptosis inhibition. J Virol. 1997;71(3):2171–2181. [PMC free article: PMC191324] [PubMed: 9032351]
- 56.
- Fessler SP, Chin YR, Horwitz MS. Inhibition of tumor necrosis factor (TNF) signal transduction by the adenovirus group C RID complex involves downregulation of surface levels of TNF receptor 1. J Virol. 2004;78(23):13113–13121. [PMC free article: PMC525002] [PubMed: 15542663]
- 57.
- Chin YR, Horwitz MS. Mechanism for removal of tumor necrosis factor receptor 1 from the cell surface by the adenovirus RIDalpha/beta complex. J Virol. 2005;79(21):13606–13617. [PMC free article: PMC1262606] [PubMed: 16227281]
- 58.
- Shisler J, Yang C, Walter B, et al. The adenovirus E3-10.4K/14.5K complex mediates loss of cell surface Fas (CD95) and resistance to Fas-induced apoptosis. J Virol. 1997;71(11):8299–8306. [PMC free article: PMC192288] [PubMed: 9343182]
- 59.
- Benedict CA, Norris PS, Prigozy TI, et al. Three adenovirus E3 proteins cooperate to evade apoptosis by tumor necrosis factor-related apoptosis-inducing ligand receptor-1 and - 2. J Biol Chem. 2001;276(5):3270–3278. [PubMed: 11050095]
- 60.
- Tollefson AE, Toth K, Doronin K, et al. Inhibition of TRAIL-induced apoptosis and forced internalization of TRAIL receptor 1 by adenovirus proteins. J Virol. 2001;75(19):8875–8887. [PMC free article: PMC114456] [PubMed: 11533151]
- 61.
- Tollefson AE, Stewart AR, Yei SP, et al. The 10,400- and 14,500-dalton proteins encoded by region E3 of adenovirus form a complex and function together to down-regulate the epidermal growth factor receptor. J Virol. 1991;65(6):3095–3105. [PMC free article: PMC240965] [PubMed: 1851870]
- 62.
- Delgado-Lopez F, Horwitz MS. Adenovirus RIDalphabeta complex inhibits lipopolysaccharide signaling without altering TLR4 cell surface expression. J Virol. 2006;80(13):6378–6386. [PMC free article: PMC1488987] [PubMed: 16775326]
- 63.
- Efrat S, Fejer G, Brownlee M, et al. Prolonged survival of pancreatic islet allografts mediated by adenovirus immunoregulatory transgenes. Proc Natl Acad Sci USA. 1995;92(15):6947–6951. [PMC free article: PMC41448] [PubMed: 7624350]
- 64.
- Efrat S, Serreze D, Svetlanov A, et al. Adenovirus early region 3(E3) immunomodulatory genes decrease the incidence of autoimmune diabetes in NOD mice. Diabetes. 2001;50(5):980–984. [PubMed: 11334441]
- 65.
- Pierce MA, Chapman HD, Post CM, et al. Adenovirus early region 3 antiapoptotic 10.4K, 14.5K and 14.7K genes decrease the incidence of autoimmune diabetes in NOD mice. Diabetes. 2003;52(5):1119–1127. [PubMed: 12716741]
- 66.
- Filippova M, Song H, Connolly JL, et al. The human papillomavirus 16 E6 protein binds to tumor necrosis factor (TNF) R1 and protects cells from TNF-induced apoptosis. J Biol Chem. 2002;277(24):21730–21739. [PubMed: 11934887]
- 67.
- Yuan H, Fu F, Zhuo J, et al. Human papillomavirus Type 16 E6 and E7 oncoproteins upregulate c-IAP2 gene expression and confer resistance to apoptosis. Oncogene. 2005;24(32):5069–5078. [PubMed: 15856013]
- 68.
- Baillie J, Sahlender DA, Sinclair JH. Human cytomegalovirus infection inhibits tumor necrosis factor alpha (TNF-α) signaling by targeting the 55-kilodalton TNF-α receptor. J Virol. 2003;77(12):7007–7016. [PMC free article: PMC156201] [PubMed: 12768019]
- 69.
- Popkin DL, Virgin HWt. Murine cytomegalovirus infection inhibits tumor necrosis factor alpha responses in primary macrophages. J Virol. 2003;77(18):10125–10130. [PMC free article: PMC224571] [PubMed: 12941924]
- 70.
- Doedens JR, Giddings TH Jr, Kirkegaard K. Inhibition of endoplasmic reticulum-to-Golgi traffic by poliovirus protein 3A: genetic and ultrastructural analysis. J Virol. 1997;71(12):9054–9064. [PMC free article: PMC230206] [PubMed: 9371562]
- 71.
- Neznanov N, Kondratova A, Chumakov KM, et al. Poliovirus protein 3A inhibits tumor necrosis factor (TNF)-induced apoptosis by eliminating the TNF receptor from the cell surface. J Virol. 2001;75(21):10409–10420. [PMC free article: PMC114615] [PubMed: 11581409]
- 72.
- Ghosh AK, Majumder M, Steele R, et al. Hepatitis C virus NS5A protein protects against TNF-α mediated apoptotic cell death. Virus Res. 2000;67(2):173–178. [PubMed: 10867196]
- 73.
- Park KJ, Choi SH, Choi DH, et al. 1 Hepatitis C virus NS5A protein modulates c-Jun N-terminal kinase through interaction with tumor necrosis factor receptor-associated factor 2. J Biol Chem. 2003;278(33):30711–30718. [PubMed: 12796506]
- 74.
- Majumder M, Ghosh AK, Steele R, et al. Hepatitis C virus NS5A protein impairs TNF-mediated hepatic apoptosis, but not by an anti-FAS antibody, in transgenic mice. Virology. 2002;294(1):94–105. [PubMed: 11886269]
- 75.
- Chung YL, Sheu ML, Yen SH. Hepatitis C virus NS5A as a potential viral Bcl-2 homologue interacts with Bax and inhibits apoptosis in hepatocellular carcinoma. Int J Cancer. 2003;107(1):65–73. [PubMed: 12925958]
- 76.
- Behrens SE, Tomei L, De Francesco R. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 1996;15(1):12–22. [PMC free article: PMC449913] [PubMed: 8598194]
- 77.
- Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3(3):221–227. [PubMed: 11875461]
- 78.
- DiPerna G, Stack J, Bowie AG, et al. Poxvirus protein N1L targets the I-kappaB kinase complex, inhibits signaling to NF-kappaB by the tumor necrosis factor superfamily of receptors and inhibits NF-kappaB and IRF3 signaling by toll-like receptors. J Biol Chem. 2004;279(35):36570–36578. [PubMed: 15215253]
- 79.
- Aoyagi M, Zhai D, Jin C, et al. Vaccinia virus N1L protein resembles a B-cell lymphoma-2 (Bcl-2) family protein. Protein Sci. 2007;16(1):118–124. [PMC free article: PMC2222835] [PubMed: 17123957]
- 80.
- Cooray S, Bahar MW, Abrescia NG, et al. Functional and structural studies of the vaccinia virus virulence factor N1 reveal a Bcl-2-like anti-apoptotic protein. J Gen Virol. 2007;88(Pt 6):1656–1666. [PMC free article: PMC2885619] [PubMed: 17485524]
- 81.
- Zhang Z, Abrahams MR, Hunt LA, et al. The vaccinia virus N1L protein influences cytokine secretion in vitro after infection. Ann N Y Acad Sci. 2005;1056:69–86. [PubMed: 16387678]
- 82.
- Shisler JL, Jin XL. The vaccinia virus K1L gene product inhibits host NF-kappaB activation by preventing I kappa B alpha degradation. J Virol. 2004;78(7):3553–3560. [PMC free article: PMC371086] [PubMed: 15016878]
- 83.
- Bradley RR, Terajima M. Vaccinia virus K1L protein mediates host-range function in RK-13 cells via ankyrin repeat and may interact with a cellular GTPase-activating protein. Virus Res. 2005;114(1-2):104–112. [PubMed: 16039000]
- 84.
- Tait SW, Reid EB, Greaves DR, et al. Mechanism of inactivation of NF-kappa B by a viral homologue of I kappa b alpha. Signal-induced release of i kappa b alpha results in binding of the viral homologue to NF-kappa B. J Biol Chem. 2000;275(44):34656–34664. [PubMed: 10934190]
- 85.
- Granja AG, Nogal ML, Hurtado C, et al. The viral protein A238L inhibits TNF-α expression through a CBP/p300 transcriptional coactivators pathway. J Immunol. 2006;176(1):451–462. [PubMed: 16365438]
- 86.
- Choi SH, Park KJ, Ahn BY, et al. Hepatitis C virus nonstructural 5B protein regulates tumor necrosis factor alpha signaling through effects on cellular I kappa B kinase. Mol Cell Biol. 2006;26(8):3048–3059. [PMC free article: PMC1446972] [PubMed: 16581780]
- Introduction
- TNF and TNF-Mediated Signaling
- PLAD Domain of TNFRs
- TNF-Mediated Diseases
- Current Anti-TNF Therapies in Humans
- Safety Issues with Current Anti-TNF Therapies
- Viral TNF Inhibitors as Alternative Therapeutics
- Viral TNF-Binding Proteins Unrelated to Host TNFRs
- Viral Proteins That Modulate TNF Receptors and Regulate Downstream Signaling
- Cell Signaling Inhibitors from Viruses That Inhibit Activation of NF-κB
- Conclusions
- Acknowledgements
- References
- Viral TNF Inhibitors as Potential Therapeutics - Madame Curie Bioscience Databas...Viral TNF Inhibitors as Potential Therapeutics - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...