

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Institute of Medicine (US) Committee to Assess the Science Base for Tobacco Harm Reduction; Stratton K, Shetty P, Wallace R, et al., editors. Clearing the Smoke: Assessing the Science Base for Tobacco Harm Reduction. Washington (DC): National Academies Press (US); 2001.

Clearing the Smoke: Assessing the Science Base for Tobacco Harm Reduction.
Show detailsThe impact of cigarette smoking on the U.S. national burden of cardiovascular disease (CVD) has been well documented. Each year, about 150,000 cardiovascular deaths are attributable to cigarette smoking, and of these, about 30,000 are attributable to environmental cigarette smoke exposure among nonsmokers (McGinnis and Foege, 1999, Taylor et al., 1992). The cardiovascular burden of smoking is amplified substantially because smoking significantly increases the risk for many types of cardiovascular morbidity: myocardial infarction (MI), sudden cardiac death, stroke, peripheral vascular disease, and abdominal aortic aneurysms (Green et al., 1993). Importantly, the risk of cardiac ischemic events is substantially and relatively rapidly reversible on cessation of smoking (U.S. DHHS, 1983).
In 1990–1994, an average of 430,700 Americans died each year of smoking-related illness. The largest portion of these deaths were cardiovascular-related illnesses. Approximately one in five deaths from cardiovascular diseases is attributable to smoking. According to the World Health Organization (WHO), 1 year after quitting, the risk of coronary heart disease (CHD) decreases by 50%, and within 15 years, the relative risk of dying from CHD for an ex-smoker approaches that of a long-time nonsmoker.
Despite this well-recognized association between smoking, and CVD, and sudden cardiac death, and the increasing amount of pathogenic information available, more needs to be known about the mechanisms of smoking-induced CVD injury. This is of particular importance because one constituent of tobacco smoke—nicotine—is currently used as short-term therapy for smoking cessation, and as discussed elsewhere in this volume, nicotine is being considered for extended use as an aid to smoking reduction. This necessitates a detailed consideration of the cardiovascular pharmacology of nicotine. The results of the Lung Health Study (Anthonisen et al., 1994) is notable in the consideration of the cardiovascular effects of long-term nicotine replacement therapy (NRT) since it was found that long-term use of nicotine gum for the purpose of smoking cessation did not result in an increased incidence of cardiovascular complications. Furthermore, it is pertinent to strategies being deployed by tobacco product manufacturers to eliminate selectively discrete constituents of tobacco and cigarettes and then market them as “safer.” This implies a knowledge of the relative contributions of a myriad of tobacco constituents to CVD, which simply does not exist. This chapter summarizes the current scientific basis of our understanding of smoking and CVD, suggests modern methodologies that might usefully be applied to enhance understanding in this area, and highlights areas for further research.
CORONARY HEART DISEASE
The incidence of coronary artery disease (CAD), including sudden cardiac death, is more than doubled in cigarette smokers as a group and is increased fourfold in heavy smokers. There is a dose-response relationship between cigarette smoking and CAD, such that the risk increases with the number of cigarettes smoked daily, the extent of inhalation, and the number of years of smoking. Cigarettes that nominally deliver less tar or nicotine have not been shown to confer any protection from ischemic heart disease.
The clearest understanding of smoking-induced ischemic heart disease emerges from integration of data from epidemiological and pathophysiologic investigations. In considering the participation of smoking in the etiology of CHD, it is useful to separate the effects of smoking on the development of atherosclerotic stenosis of the coronary arteries from its effects on the process that converts coronary atherosclerosis to acute coronary events.
Acute coronary events represent an abrupt transition from stable chronic CAD to one of the major consequences of ischemia: unstable angina, myocardial infarction, and sudden cardiac death. Abundant evidence supports the concept that the rupture of a lipid-rich atherosclerotic plaque with attendant thrombus formation is the initiating event in the development of the vast majority of these ischemic syndromes (Davies and Thomas, 1984; Oates, 1989).
Coronary Atherosclerosis
Age is a powerful predictor of coronary atherosclerosis, and angiographic studies indicate that the relative risk for coronary stenosis associated with having previously smoked is greatest in the youngest age groups, suggesting an acceleration of the process by smoking. Some of the best evidence is provided by a prospective autopsy study performed in Hawaii (Reed et al., 1987) and from the study of autopsies on young men who died violently (PDAY Research Group, 1990). These studies indicate that there is a strong dose-related association of smoking with atherosclerosis of the abdominal aorta, as well as an association with atherosclerosis of the coronary arteries that is significant but less robust. From these two autopsy studies, as well as from an overview of all relevant studies, one gains the impression that the significant increase in fibrous atherosclerotic plaques and atheroma of the coronary arteries is not of sufficient magnitude to account fully for the greater increase in acute coronary events that are linked to cigarette smoking.
The finding that the increase in involvement of the coronary arteries with atherosclerosis is of small magnitude is in agreement with the prospective epidemiological evidence that stable angina pectoris is increased modestly, if at all, in cigarette smokers over age 40. In men less than 40 years of age, an increase in stable angina has been detected (twofold), but at 40 years and older the risk is increased only slightly, even after adjusting the incidence of stable angina for the loss of persons at risk owing to acute coronary events (Dawber, 1980). However, excess smoking-attributable mortality rates due to heart disease continue to increase with age and smoking duration, suggesting that smoking continues to be an important independent risk factor in the elderly (Burns, 2000).
Acute Coronary Events
Prospective epidemiological studies have consistently demonstrated a substantial increase in acute coronary ischemic events in individuals who smoke (U.S. DHHS, 1983). The pernicious effect of cigarette smoking on MI and sudden cardiac death is seen at least to age 70, but the increase in risk for a given individual appears to be greatest during middle age. The largest body of prospectively acquired North American data indicates that middle-aged men who smoke have a tenfold greater risk of sudden cardiac death and a 3.6-fold increased risk of MI (Kannel et al., 1984). The risk for sudden cardiac death is disproportionately greater than that for MI, a finding that is replicated to varying degrees in other studies. Of all the coronary risk factors, cigarette smoking is the strongest predictor of sudden cardiac death. The risk for both of these acute coronary events (MI and sudden death) clearly exceeds that for stable angina. Although the incidence of coronary deaths in women during middle age is lower than that in men, cigarette smoking accounts for about half of these deaths. Pipe and cigar smoking are also associated with increased rates of heart attack rates (Nyboe et al., 1991) and coronary mortality, as well as stroke occurrence.
In most studies that have explored the relationship of cigarette smoking rates to CVD outcomes, a dose-response has been observed. Cumulative exposures in epidemiological studies are not easy to determine because of interindividual short- and long-term variations in smoking patterns and choices of products. Also, individual inhalation depth and retention may vary in order to titrate the delivery of certain levels of nicotine (Hee et al., 1995). However, population studies employing even a relatively simple measure of exposure, such as cigarettes per day, usually reveal increasing adverse CVD outcomes with increasing numbers of cigarettes consumed. An example is the Cancer Prevention Study (CPS), shown in Table 13–1. Here, although some cells have small numbers of participants, there is a general trend of increased risk of CAD death with increasing baseline cigarette usage, most prominent up to 20 cigarettes per day.
TABLE 13–1
Age-Specific and Age-Adjusted Death Rates From Coronary Heart Disease, by Number of Cigarettes Currently Smoked.
Cessation of Smoking
Smoking cessation in individuals without known coronary heart disease makes an important contribution to reducing the risk of MI (Cook et al., 1986; Rosenberg et al., 1985, 1990; U.S. DHHS, 1983). The excess risk of MI falls by about 50% within the first two years after cessation of smoking, consistent with a substantial component of the risk of acute ischemic events being reversible. An example of the rate of decline in risk of fatal CAD, total CAD, and nonfatal MI is shown in Table 13–2 from the CPS (Stellman and Garfinkel, 1986). For those who have ceased smoking for 10–14 years, their general CVD risk declines to that of “never smokers.” A similar or even more accelerated risk reduction can be seen for stroke in women, as shown in Figure 13–1 (Kawachi et al., 1993). Even among individuals over 60 years of age, a decrease in risk for ischemic heart disease postcessation has been found, though smaller than for younger individuals (Burns, 2000).
TABLE 13–2
Time Since Quitting and Age-Adjusted and Multivariate RRs of Fatal Coronary Heart Disease and Nonfatal Myocardial Infarction, Compared with Current Smokers.
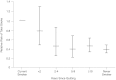
FIGURE 13–1
Risk of total stroke by time since quitting. NOTE: Age-adjusted relative risk of total stroke in relation to time since stopping smoking. Current smoker was the reference category. Error bars represent 95% confident intervals.
For smokers who already have CAD, cessation is also very effective in reducing the incidence of further acute coronary events. Survivors of MI have a greater risk of reinfarction, and survivors of sudden cardiac death have a greater risk of sudden death if they continue to smoke. For individuals who have angina pectoris or a positive exercise test or who have had coronary artery bypass graft surgery, continuing the smoking habit confers a worse prognosis. Cessation of smoking after coronary angioplasty or vascular surgery reduces the rate of restenosis by one-third. Thus, there is a major incentive for smoking cessation in this group of patients. When efforts to cease smoking are effective, they probably confer a greater benefit than any pharmacological or surgical intervention aimed at coronary disease.
Overall, these observations are encouraging for the motivation to quit. The decline of risk upon smoking cessation can be variable in different risk groups and according to behaviors during cessation, potentially affording insight into interindividual differences in mechanisms relevant to healing smoking-induced cardiovascular injury. An initial interpretation of these data suggested that acute factors, such as hemostatic activation, dominated over more gradual processes, such as occlusive atherosclerotic disease contributed to the smoking-induced CVD burden. It is now appreciated that anatomic atherosclerosis may also restructure toward more normal vessels at rates comparable to the clinical and epidemiological findings, bringing vascular factors relevant to plaque stability into consideration. In summary, the kinetics of declining CVD risk after smoking cessation do not serve to discriminate easily between hemostatic and other vascular factors as mediators of smoking-induced disease.
However, the pattern of decline in CVD risk after quitting suggests that CVD may offer a particular advantage in assessing tobacco-related harm reduction over a reasonable time. That is, patterns of change in morbidity and mortality rates after changes from a conventional to a risk reduction tobacco product may help reveal whether lower toxic exposures are actually being achieved and whether they lead to altered CVD outcomes, without waiting the decades necessary to evaluate the outcomes of smoking these products beginning from initiation of the smoking habit. The issues of compensation, nicotine dose-response relationships, and aspects of nicotine-related cardiovascular risk are discussed in Chapter 9.
Smoking and Other Cardiac Risk Factors
Cigarette smoking, hypercholesterolemia, and hypertension have been the most extensively studied risk factors for CAD. The risk of CAD imposed by cigarette smoking is magnified by the presence of several other factors that cause coronary heart disease. Cigarette smoking alone imposes a risk for CHD that is independent of other risk factors. However, smoking in conjunction with another risk factor increases the actual rate of coronary heart disease events to a greater extent than smoking alone, and these risk factors may work additively or geometrically. In one large study (The Pooling Project Research Group, 1978), smoking increased the ten-year rate of a first CAD event (MI or sudden cardiac death) by 31 per 1,000 persons when neither hypercholesterolemia nor hypertension was present. In conjunction with either hypercholesterolemia or hypertension, cigarette smoking increased the rate by 49 per 1,000 persons, and when both hypercholesterolemia and hypertension were present, the superimposition of smoking increased the rate by 97 per 1,000 persons. In women, there is a tenfold increase in the risk of MI among oral contraceptive users who smoke. Therefore, hypercholesterolemia, hypertension, and oral contraceptive use provide an incentive to not smoke or to cease smoking that exceeds the abundant benefit of avoiding this addiction in the rest of the population. Further, control of elevated cholesterol and high blood pressure reduces the risk of CAD (Brown, 2000; Mormando, 2000; The sixth report, 1997), as does smoking cessation.
The pathophysiological impact of various environmental exposures and risk factor states may be modified by host genetic factors. For example, a certain polymorphism in coagulation Factor V (Q506) confers resistance to interaction with the endogenous antithrombotic protein activated protein C. Following presentation with an acute coronary syndrome, smokers with this polymorphism have an increased risk of MI and death compared to smokers with a normal genotype, extending to at least two years after the initial event (Holm et al., 1999). Another example is the interaction between smoking and the guanosine-adenosine at the G455A polymorphism in the b-fibrinogen gene. This polymorphism is associated with higher levels of circulating fibrinogen, which in turn have been associated with increased risk of CAD (Humphries et al., 1999). This association is more pronounced in smokers (Green et al., 1993; Humphries et al., 1999). Similarly, polymorphisms in the coagulation factor VII gene have been associated with reductions in risk of CAD (Donati et al., 2000). Cigarette smoking interacts to a variable degree with genes related to other aspects of cardiovascular risk, including antioxidant enzymes such as paraoxonase (Sen-Banerjee et al., 2000), apoliprotein B (Glisic et al., 1995), DNA repair genes (Abdel-Rahman and El-Zein, 2000), and proteins that regulate the availability of cardiotoxic autacoids such as the serotonin transporter (Arinami et al., 1999). Finally, the cardiovascular hazards of tobacco may be modulated by the presence of functionally important polymorphisms in enzymes that detoxify harmful constituents of tobacco smoke (Li et al., 2000). Clearly, emerging information about genetic polymorphisms that are of functional significance and relevance to the cardiovascular system will afford an increasing opportunity to understand and predict the effect of smoking on CVD risk at the individual level.
Environmental Tobacco Smoke
There is considerable evidence that exposure to environmental tobacco smoke (ETS; passive smoking) has an adverse effect on cardiovascular health (Kawachi et al., 1997a). Pooled results of epidemiological studies indicates a 20% excess coronary heart disease death rate among nonsmoking spouses of smokers (Steenland et al., 1996). As many as 40,000 deaths from MI each year may be the result of passive smoking. The mechanisms linking ETS exposure and cardiovascular disease are probably similar to those for active smoking.
Pathophysiology
A review of the mechanisms linking cigarette smoking and coronary heart disease must consider the fact that whereas smoking accelerates coronary atherosclerosis, it has an even greater effect on the process that abruptly converts atherosclerosis to the acute ischemic events of MI, unstable angina, and sudden cardiac death. The effect of chronic smoking on the initiation of acute ischemic events appears to be largely reversible, given the substantial and rapid reduction in the incidence of acute ischemic events after smoking cessation. Although there is likely overlap between the mechanisms by which smoking accelerates atherosclerosis and promotes acute ischemic events, it is useful to consider these adverse effects of smoking separately.
Acceleration of Atherosclerosis
As for other risk factors, vascular endothelial dysfunction likely plays a central role in the promotion of atherosclerosis in chronic smokers (FitzGerald et al., 1988; Heitzer et al., 1996a). Extensive endothelial abnormalities are present in the umbilical arteries of infants born to smoking mothers, and lesions of endothelial cells, subendothelial damage, and platelet adhesion have been described in vessels of experimental animals exposed to cigarette smoke. Increased in vivo prostacyclin biosynthesis and functional abnormalities of vascular endothelium also result from cigarette smoking. Impaired endothelium-dependent vasodilatation in both forearm and coronary vascular beds has been demonstrated even in young smokers (Campisi et al., 1998; Heitzer et al., 1996b; Zeiher et al., 1991). These functional abnormalities may be due to smoking-induced inhibition of nitric oxide (NO) release (Campisi et al., 1999; Kugiyama et al., 1996) or to acceleration of NO breakdown. Structural endothelial damage may result either from a direct toxic effect of nicotine or other components of cigarette smoke on endothelial cells or from smoking-induced oxidative stress (Heitzer et al., 1996a, b). Smokers have reduced levels of antioxidant vitamins and increased levels of oxidized low-density lipoprotein (LDL), a potent inhibitor of endothelial function (Heitzer et al., 1996b; Morrow et al., 1995). Smoking-induced acute and chronic systemic hemodynamic changes may also contribute to vascular endothelial dysfunction. For example, some but not all studies suggest that smoking a single cigarette causes an acute rise in systemic blood pressure and heart rate, and chronic smoking results in a persistent elevation in daytime blood pressure. The effect of chronic smoking on blood pressure is more complex as outlined later.
Smoking is associated with lipid abnormalities that may contribute to the development of atherosclerosis (Duthie et al., 1993; Craig et al., 1989). In addition to increased levels of oxidized low-density lipoprotein, smoking produces an increase in very low density lipoprotein (VLDL) and triglycerides and a reduction in high-density lipoprotein (HDL) levels. Smoking has been shown to increase monocyte adhesion to endothelial cells, an important early event in atherosclerosis (Adams et al., 1997; Weber et al., 1996). Additional factors that may contribute to the development of coronary and other atherosclerotic events in smokers include smoking-induced platelet activation, increased fibrinogen levels, and increased blood viscosity.
Promotion of Acute Coronary Events
In addition to the effect of smoking on the development of atherosclerosis, there is a dramatically increased risk of acute ischemic events, including MI and sudden death. Acute systemic and coronary hemodynamic effects of smoking are likely to play an important role in the development of these ischemic events. After smoking a single cigarette, systemic arterial pressure, heart rate, and myocardial contractility increase, resulting in a rise in myocardial oxygen demand (Cryer et al., 1976; Nicod et al., 1984). Simultaneously, in patients with atherosclerosis, smoking causes acute vasoconstriction of both conduit and resistance coronary vessels, with a decrease in coronary blood flow (Czernin et al., 1995; Nicod et al., 1984; Quillen et al., 1993). Even in the absence of a hemodynamically significant stenosis, coronary flow may fall by more than 20% despite a significant increase in myocardial oxygen demand. In some individuals, smoking causes intense focal vasoconstriction or spasm that can lead to myocardial ischemia (Moauad et al., 1986). These acute hemodynamic effects of smoking in the coronary bed are most likely adrenergically mediated, since they can be prevented by α-adrenergic blockade (Winniford et al., 1986). Adrenergic stimulation has been shown to cause exaggerated coronary vasoconstriction in the setting of endothelial dysfunction (Vita et al., 1992; Zeiher et al., 1989). Plasma norepinephrine and epinephrine levels rise acutely after smoking (Cryer et al., 1976); this catecholamine release may lower arrhythmia threshold and increase the risk of sudden death.
In addition to lowering anginal threshold, repeated episodes of coronary vasoconstriction and elevations of systemic arterial pressure may increase hemodynamic stresses at the site of an atherosclerotic plaque. An increase in shear stress at the site of a vulnerable plaque is considered a potentially important cause of plaque rupture.
Smoking has also been shown to exacerbate the cardiovascular effects of cocaine (Moliterno et al., 1994). Both the epicardial coronary constriction and the increase in myocardial oxygen demand caused by cocaine are potentiated by concomitant smoking, perhaps increasing the risk of MI and sudden death attributed to cocaine use.
Inhaled carbon monoxide (CO) in cigarette smoke binds to hemoglobin, reducing oxygen delivery to myocardial cells. The carbon monoxide levels found in smokers have been shown to lower anginal threshold (Allred et al., 1989; Aronow, 1981) and increase ventricular fibrillation threshold (Aronow et al., 1979). Carbon monoxide may also have a deleterious effect on vascular endothelial cells (Thom et al., 1997).
Coronary thrombosis plays a major role in acute coronary events, and smoking is associated with several changes in the hemostatic system that lead to a hypercoagulable state. These hemostatic changes include an increase in fibrinogen (De Maat et al., 1996), red cell mass, and blood viscosity; platelet activation with thromboxane A2 release (Benowitz et al., 1993); and impaired release of tissue plasminogen activator from endothelial cells (Newby et al., 1999).
Most research on the effects of smoking on the cardiovascular system has focused specifically on nicotine and carbon monoxide, but there are more than 4,000 other components in cigarette smoke, some of which may also contribute to smoking-induced vascular disease (Penn and Snyder 1996).
Coronary Vasospasm
A much less common cause of ischemia than plaque rupture, primary coronary vasospasm usually manifests as variant angina but may also cause MI and sudden cardiac death. Coronary vasospasm is strongly associated with cigarette smoking (Okasha et al., 2000).
EXTRACARDIAC VASCULAR DISEASE
Smoking markedly accelerates atherosclerosis in the abdominal aorta, and occlusive disease of its branches is increased as well (Reed et al., 1987; PDAY, 1990). Aortic aneurysm, peripheral vascular disease, and renal artery stenosis are increased in smokers. Renovascular hypertension should be strongly suspected in cigarette smokers who have severe hypertension. Cigarette smoking is an independent risk factor in the development of atherosclerosis in the internal pudendal and penile arteries of young men with impotence.
Two types of stroke are increased in cigarette smokers (U.S. DHHS, 1989). Smoking is a risk factor for cerebral infarction, and the magnitude of risk increases with greater smoking exposure. This occurs in conjunction with an increase in atherosclerosis of the carotid arteries in smokers. Subarachnoid hemorrhage is markedly increased in cigarette smokers, and in women who take oral contraceptives, this risk if further enhanced.
OTHER VARIABLES INFLUENCED BY SMOKING
Blood Pressure
The effect of cigarette smoking on chronic blood pressure levels in uncertain, in part because of concomitant effects of lower body mass index (BMI) and more frequent alcohol consumption among smokers. Some studies have reported lower ambulatory blood pressure levels (Mikkelsen, et al., 1997; Okasha et al., 2000), while others have reported increases (Minami et al., 1999; Mundal et al., 1997). Findings may depend in part on when in the day pressure recordings are made and whether smoking is assessed by self-report or by biochemical verification (Istvan et al., 1999). Smoking in healthy volunteers has been reported to alter the change in blood pressure response to changes in body position (Nardo et al., 1999) and to cause abnormal baroreceptor function (Gerhardt et al., 1999). Of note, maternal smoking has been associated with increased blood pressure levels in the children of these pregnancies (Blake et al., 2000).
Insulin Resistance
The insulin resistance syndrome, as well as insulin resistance per se, has been associated with increased risk of atherosclerotic vascular disease, independent of most other major risk factors and is a risk factor for non-insulin-dependent diabetes mellitus (NIDDM) (Mikhailidis et al., 1998). Increased insulin resistance has been associated with increased intimal-medial thickening in the presence of vasospastic angina (Shinozaki et al., 1997). The syndrome occurs in healthy young men and is sixfold more common in cigarette smokers (Tahtinen et al., 1998). Insulin resistance is also increased among smokers with existing NIDDM (Targher et al., 1997). Smoking cessation improves insulin sensitivity among healthy middle-aged men (Eliasson et al., 1997).
Other Lipid or Lipoprotein Measures
Lipoprotein (a) (Lp(a)) cholesterol levels are positively and independently correlated with the risk of coronary events in some but not all prospective studies (Seman et al., 1999). Cigarette smoking does not appear to alter blood Lp(a) levels (Chien et al., 1999; Kamboh et al., 2000). However, smoking appears to alter the ratio of LDL subfractions in the direction associated with increased CVD risk (Griffin et al., 1994).
Inflammation
There is substantial evidence that inflammation plays a role in the pathogenesis of atherosclerotic lesions in many parts of the arterial tree and that elevated blood levels of cytokines such as interleukin-6 (IL-6) and C-reactive protein (CRP) are associated with increased risk of coronary events (Ridker et al., 2000; Koenig et al., 1999). Smoking is associated with increased levels of CRP, suggesting that inflammation is one mechanism by which smoking promotes the pathogenesis of these lesions. Smoking has also been shown to increase local tissue levels of some cytokines in organs other than the vascular tree, such as tumor necrosis factor-alpha (TNF-α) in gingival cervicular fluid, but not IL-6 (Bostrom et al., 1999) or IL-8 and ICAM-1 in small respiratory airways (Takizawa et al., 2000).
Homocysteine
Blood homocysteine levels have been related to an increased risk of coronary heart disease and other atherosclerotic diseases in several studies (Saw, 1999). Cigarette smoking has been shown to be associated with higher blood homocysteine levels, both in the basal state and after a methionine load (Reis et al., 2000; Nygard et al., 1998). In a study in which the dose-response was explored, there was a clear positive correlation between the number of cigarettes smoked and blood homocysteine levels. This raises the possibility that smoking cessation may have its effects in part through this mechanism and that homocysteine level might be developed as a biomarker of smoking exposure.
SURROGATE MARKERS
The identification and validation of quantitative indices that reflect biochemical mechanisms of cardiovascular injury caused by cigarette smoking would be useful. Such surrogate indices might permit the study of both progression and regression of smoking-related cardiovascular disease and clarify the factors that contribute to interindividual differences in susceptibility to such complications. They might also be useful correlates of the relative bioavailability of individual constituents of cigarette smoke and aid in the evaluation of harm reduction strategies, including potentially “safer” smoking devices and nicotine replacement therapies. They might also prove of value in assessment of the cardiovascular risk of secondhand smoke. Presently, little information is available relating to the mechanisms of cigarette-induced cardiovascular damage, but the emergence of promising technologies relating to potentially important mechanisms, including platelet activation and oxidative stress, illustrates the potential of the approach.
Cigarette smoking causes platelet activation in vivo, as reflected by an accelerated platelet turnover time (Fuster et al., 1981). However, the development of noninvasive approaches to assessment of platelet activation facilitates repeated investigation of the effects of smoking on platelet function. Platelets are extremely susceptible to platelet activation ex vivo during blood sampling, and this has constrained the usefulness of circulating biomarkers of platelet activation, such as circulating platelet aggregates and β-thromboglobulin or, indeed, of more contemporary approaches such as P selectin expression on platelets ex vivo. Studies of the platelet aggregation response ex vivo have also proven uninformative. Platelets activated by cigarette smoke in vivo might be less available for harvesting for such ex vivo studies. Indeed, platelet responses to adenosine 5′-diphosphate (ADP) were reportedly diminished ex vivo in smokers in one large-scale epidemiological study (Meade et al., 1985). By contrast, urinary excretion of the 2,3-dinor and 11-hydro metabolites of thromboxane have proven to be noninvasive indices of platelet activation in vivo (FitzGerald et al., 1983). Thus, phasic increases in excretion of these metabolites occur during the ischemic episodes of unstable angina and in patients undergoing therapeutic thrombolysis (Fitzgerald et al., 1988; Lewis et al., 1983), two situations in which the functional importance of thromboxane dependent platelet activation is implied by the therapeutic efficacy of aspirin (Intravenous streptokinase, 1987; Lewis et al., 1983).
Excretion of thromboxane metabolites is increased in apparently healthy individuals who smoke and decreases on short-term cessation (Nowak et al., 1987). This observation has been confirmed, both in a large population study and in a case-control study of monozygotic twins discordant for the smoking habit (Lassila et al., 1988; Wennmalm et al., 1991). Interestingly, excretion of the major metabolite of prostacyclin, 2,3-dinor-6-keto-PGF1α (prostaglandin F1α) (Brash et al., 1983), is also elevated in chronic cigarette smokers. Prostacyclin, like thromboxane, is a product of cyclooxygenase (COX) catalyzed metabolism of arachidonic acid. While prostacyclin is the major product of COX in endothelial cells, thromboxane is the principal product of the same enzyme in platelets. Platelets express exclusively COX-1, and the suppression of thromboxane biosynthesis by low-dose aspirin (Nowak et al., 1987) implies that this is the major source of its formation in smokers (Lassila et al., 1988). However, COX-2 in the vessel wall or in infiltrating cells, such as monocyte or macrophages, represents a second theoretical source of thromboxane formation. Indeed, COX-2 seems the likely source of prostacyclin formation (Wennmalm et al., 1991; Brash et al., 1983; McAdam et al., 1999), although the effects of selective inhibitors of COX-2 on the formation of either eicosanoid in cigarette smokers remains to be reported. Although cigarette smoke reduces the capacity of endothelial cells to form prostacyclin in vitro (Reinders et al., 1986), the capacity of cells to form this and other eicosanoids greatly exceeds actual biosynthetic rates in vivo (FitzGerald et al., 1983). The increment in prostacyclin biosynthesis coincident with that of thromboxane is consistent with the capacity of thromboxane analogues to evoke its formation by endothelial cells in vitro (Nicholson et al., 1984) and its role as a homeostatic response to accelerated platelet vessel wall interactions in vivo (FitzGerald et al., 1984). Deletion of the prostacyclin receptor increases the response to thrombotic stimuli (Murata et al., 1997). While an abundant literature supports the use of these indices of platelet-vascular interactions, they could be usefully related to other, more functional surrogates of the cardiovascular response to smoking, such as impaired endothelial function (Raitakari et al., 2000) and noninvasive assessment of atherosclerosis progression, using ultrasonography or electron beam computerized tomography (EBCT) (O'Malley et al., 2000; Raggi et al., 2000). Such surrogates could represent an intermediate link between the noninvasively acquired biochemical indices and true functional outcomes of smoking, such as myocardial infarction and sudden death.
Another mechanism that has been widely implicated in cigarette-induced tissue injury is oxidative stress. Free-radical-catalyzed damage has been suggested to be of relevance to both atherogenesis and tumor progression and, as such, may well be relevant to the harm caused by cigarette smoking. Indeed, cigarette smoke contains abundant radicals, and smoking is known to cause activation of platelets and neutrophils, which may in turn increase radical generation (Pryor, 1997; Shinagawa et al., 1998; Yamaguchi et al., 2000). Until recently, the study of oxidative damage in vivo has been constrained by the specificity of available biomarkers. However, the discovery of isoprostanes (iPs), free-radical-catalyzed products of arachidonic acid (Lawson et al., 1999; Roberts and Morrow, 2000), has transformed this situation. These products are formed initially in situ in the phospholipid domain of cell membranes, where they may be immunolocalized or quantitated directly (Roberts and Morrow, 2000). Following phospholipase-dependent cleavage, iPs circulate and are excreted in urine. Urinary iPs reflect the oxidant stress attendant on inflammation (McAdam et al., 2000) and are increased in a dose-dependent manner in cigarette smokers (Reilly et al., 1996). Some evidence exists suggesting their elevation by secondhand smoke and in the children of smokers (Sinzinger et al., 2000). Elevated urinary iPs in smokers appear to fall rapidly on quitting (McAdam et al., 2000; Morrow et al., 1995; Reilly et al., 1996; Sinzinger et al., 2000). It is possible that iPs may be of functional relevance to smoking-induced cardiovascular injury besides serving as a marker of the process. Thus, individual iPs may act as incidental ligands both at membrane receptors for prostanoids (Kunapuli et al., 1998; Morrow et al., 1995) and at peroxisomal proliferator activated receptors (PPARs) (Audoly et al., 2000). Thus, certain iPs activate platelets via thromboxane receptors in smokers (McNamara et al., 1999) and may, in turn, lead to generation of thromboxane itself, creating a vicious cycle (Davi et al., 1999). In this setting, effective antioxidants might suppress both iP generation and thromboxane biosynthesis. Cigarette smokers are susceptible to depletion of endogenous vitamin C, and supplementation of smokers with vitamin C depresses elevated iPs (Valkonen and Kuusi, 2000). By contrast, their endogenous levels of vitamin E are replete, and exogenous vitamin E does not suppress elevated iPs in smokers (Reilly et al., 1996). Other noninvasive indices of lipid peroxidation, such as 4-hydroxynonenal (Benedetti et al., 1980), and of radical-induced protein (Pennathur et al., 1999) and DNA (Marnett, 2000) damage have begun to emerge in clinical investigation. No information is available on relative effects of these distinct targets for radical-induced damage or their relevance to smoking-induced cardiovascular injury.
In summary, the examples of urinary thromboxane, prostacyclin, and isoprostanes illustrate the potential value of developing and validating quantitative, noninvasive biochemical indices of the mechanisms of smoking-induced cardiovascular damage and tissue injury in general. Initially, the methodology involved (e.g., mass spectrometry) is often expensive and labor intensive. However, once the proof of principle for such approaches has been established, effort can be rationally devoted to the development of simpler, more widely applicable, but validated methodologies, such as enzyme immunoassays. The power of this approach, which can also be applied to animal models (Pratico et al., 1998; Heinecke, 1999), will be greatly enhanced by its integration with surrogate markers of functional impairment and, ultimately, with actual clinical events in the assessment of the cardiovascular risks of smoking.
Potential surrogate markers to be used in animal models developed to detect potentially adverse effects of smoking and of the use of potential reduced-exposure products (PREPs) on arteries throughout the body and in the lungs and heart include measurements of platelet activation, blood pressure and heart rate, and oxidative stress. Other surrogate markers that may be utilized in clinical studies include measurements of oxidative stress, measurements of platelet activation, elements of the coagulation system, lipids and lipoproteins, pulmonary function studies, studies of endothelial function, myocardial stress perfusion studies, and periodic evaluation by ultrasound of the carotid, abdominal aortic, and lower-extremity arteries.
CONCLUSIONS
Dose-Response Relationship
Highly informative information on the existence of a dose-response relationship between cigarette exposure and cardiovascular risk comes from many studies such as the CPS-2 (Thun et al., 1997) and Harvard Nurses' Health Studies (Kawachi et al., 1997b). In both instances, there is a relationship between the number of cigarettes smoked and the incidence of cardiovascular events. This is illustrated for the incidence of myocardial infarction (Table 13–3) and stroke (Table 13–4). In both cases, the most striking difference is between nonsmokers and individuals who smoke the least number of cigarettes recorded. The relationship becomes somewhat less pronounced as the number of cigarettes smoked per day increases. Interestingly, ex-smokers tend to occupy a space intermediate between nonsmokers and those with the lowest daily smoking frequency.
TABLE 13–3
Number of Cigarettes Smoked Daily and Age-Adjusted and Multivariate RRs of Fatal Coronary Heart Disease and Nonfatal Myocardial Infarction, Compared with Never Smokers.
TABLE 13–4
Age-Adjusted RRs of Stroke (Fatal and Nonfatal Combined), by Daily Number of Cigarettes Consumed Among Current Smokers.
This dose-response curve prompts several considerations. First, there is no persuasive evidence of a threshold below which a cardiovascular risk does not exist. This observation affirms the primary objective of encouraging smokers to quit completely. Second, the shallow dose-response relationship, with the impression of a plateau, accords with similar observations relating the number of cigarettes smoked and measurements of systemic bioavailability, such as nicotine and cotinine. The same is true of the relationship with CO (Gori and Lynch, 1985). Although this may represent saturation kinetics of nicotine or CO, the most likely explanation is compensation for lower numbers of cigarettes smoked. Thus, the smoker titrates nicotine delivery toward a range of convergence that is reflected by the measurement of nicotine delivery, which in turn, may reflect dose-dependent convergence of the delivery of additional toxic, but unmeasured, constituents of cigarette smoke. The relative contribution of distinct constituents of cigarette smoke to smoking-related cardiovascular morbidity and mortality is unknown. In this regard, the available information is incomplete regarding any differences between the dose-response relationship of filter or low-tar versus high-tar or unfiltered cigarettes.
Feasibility of Harm Reduction in Therapy
There are no data that are directly informative on the issue of harm reduction. Thus, although a dose-response relationship exists for cardiovascular and cerebrovascular events, we do not know if reducing the number of cigarettes smoked results in a quantitative reduction in the risk of these events. However, the intuitively appealing prospect that this is indeed the case is supported by evidence from individuals who quit smoking. Thus, quitting results in a time-dependent reduction in the incidence of myocardial infarction (Table 13–2) and stroke. The latter is most nicely illustrated by data relating to subarachnoid hemorrhage, which was significantly elevated in women smokers in the Nurses' Health Study (Figure 13–1). In addition to evidence from such unequivocal clinical events, there is evidence that the increase in biomarkers of oxidant stress, platelet activation, and inflammation (Benowitz et al., 1993), all of potential mechanistic relevance to tobacco-related cardiovascular injury, rapidly falls toward the normal range on quitting cigarettes. The offset kinetics of more functional surrogates, such as endothelial dysfunction, remain to be determined in smokers. In summary, the data from quitters encourage the prospect that a graded reduction in cardiovascular risk and in biomarkers of this risk may accompany a reduction in the number of cigarettes smoked in pursuit of a harm reduction strategy. It is possible, indeed likely, that individuals and perhaps populations, differ in their susceptibility to tobacco-induced cardiovascular risk and, indeed, in their potential benefit from a harm reduction strategy. Data from quitting studies indicate a considerable variance in the rate of offset of risk, which declines with time. No data are available to address such issues across ethnic groups or gender. Acquisition of such information and research on the environmental and genetic factors that condition interindividual variability in exposure-risk relationships are necessary.
Utility in a Preclinical Setting
Studies in cell culture and model systems can afford much needed information on tobacco-related cardiovascular risk. These might include a profiling of gene expression and translation in cardiovascular tissues in response to cigarette smoke, constituents of smoke, and potential risk reduction substituents. These might identify proteins of potential functional relevance to the transduction of cardiovascular risk. Such studies might be coupled with gene inactivation and overexpression studies to address the role of these proteins in vivo. Similarly, studies of exposure to cigarette smoke or to discrete constituents of smoke might be deployed to investigate effects on atherosclerosis progression, susceptibility to vascular injury, thrombotic stimuli, graft rejection, cardiovascular development, or endothelial dysfunction in model systems such as mice. Studies of cardiovascular genomics and ultimately proteomics can also be extended to model systems to investigate gene expression and translation in response to exposure to tobacco-related products in vivo. These observations may, in turn, be related to the pattern of gene expression and translation in cardiovascular tissues obtained from cigarette smokers.
Biomarkers of Tobacco-Related Disease
The predominant mechanisms by which cigarette smoking induces cardiovascular injury is unknown. However, small studies in smokers of potentially relevant biomarkers of platelet and vascular activation, lipid peroxidation, and inflammation afford evidence of a dose-response relationship and a decline on quitting. There is even evidence of a signal in individuals exposed to ETS in the case of some of these markers. More mechanism-based clinical studies are required to confirm and expand these findings. Where possible, these should be related to surrogate measurements of cardiovascular function, such as hemodynamics, flow-mediated endothelial function and estimates of plaque progression by ultrasound or EBCT. Furthermore, biomarker studies can usefully be integrated into many studies in model systems (see above) as well as studies of clinical outcome (discussed below) to afford their ultimate validation.
Clinical Assessment of Tobacco-Related Disease
The time course of offset of myocardial infarction and stroke in people who stop smoking suggests that cardiovascular disease represents a tractable scenario in which one might evaluate harm reduction strategies. Clearly, assessment of the impact of such events can occur in a fairly reasonable time compared to that which is possible for cancer or lung disease.
RESEARCH AGENDA
- 1.
Determine whether physiological and biochemical measures that are altered by tobacco products may be used as indicators of disease risk and as a means to distinguish various forms of tobacco use in a broad range of exposures (doses), including:
- activation of platelet function;
- endothelial function;
- endothelial thickening and plaque formation;
- blood pressure and heart rate alterations;
- vascular aneurysm formation;
- vascular thrombosis (arterial and venous);
- cardiac electrophysiology, including ventricular and atrial ectopy, heart rate variability, and variations in conduction times, especially QT intervals; and
- other risk factors for cardiovascular disease including lipid and carbohydrate abnormalities, homocysteine, folic acid and antioxidant vitamins, markers of inflammation, and immune function.
Based on current evidence, oxidative stress seems likely to play an important role in mediating the cardiovascular effects of smoking. Determining reliable biomarkers of oxidative stress and the interaction of unstable oxygen intermediates with lipids, proteins, and DNA should receive high priority. Since inflammation is a proximate cause of oxidative stress, further work on biomarkers of inflammation such as C-reactive protein, cytokines, and acute-phase proteins and the effects of tobacco exposure appears to be an important research direction. Similarly, further assessment of the effects of various tobacco products on biomarkers of hemostatic activation and platelet function should be fruitful in understanding and predicting atherogenesis.
- 2.
Develop studies to determine whether new biomarkers, such as products of lipid peroxidation and corresponding adducts to DNA, and products of arachidonic acid metabolism can serve as intermediate indicators (risk factors) of cardiovascular disease risk related to different tobacco products, and tobacco-related PREPs in particular. Through well-designed clinical trials, determine the ability of antioxidant or related therapies to counter the tobacco-related perturbations due to oxidative stress that are likely to be caused by both conventional and new tobacco products.
- 3.
Develop long-term observational studies in individuals exposed to conventional and modified tobacco products primarily and secondarily (i.e., passive smoking), better to identify actual risks for cardiovascular disease, including dose-response characteristics. Included should be evaluations of the variables described in items 1 and 2, subsequent cardiovascular disease development, and potential differences in different tobacco or smoking products.
- 4.
Further apply modern in vitro and other model systems, and ex vivo in humans, to assist in elucidating the comparative toxicology or adverse effects of cigarette smoke and PREPs. Such studies might include the use of gene array and proteomic technologies to study differential gene expression and translation; the use of chronic exposures to investigate effects on atherosclerosis progression, response to thrombotic and arrhythmogenic stimuli, and cardiovascular disease development; and studies of the effects of cigarette smoke and its constituents on the function and integrity of cardiovascular cells in vitro, including platelets, leukocytes, endothelial cells, vascular smooth muscle cells, and myocytes.
- 5.
Apply modern genomic and proteomic analyses to elucidate the array of genes of potential relevance to cardiovascular hazards differentially induced by exposure to conventional tobacco products and PREPs. Utilize such techniques as transgenic and knockout model systems to elucidate the relevance of single genes to vascular injury induced by cigarette smoke and its discrete constituents, including nicotine. Assess the possibility of allelic variation in such genes and its relevance to smoking-induced cardiovascular disease risk.
- 6.
Continue to elucidate further aspects of the vascular biology of nicotine in view of its effects on flow-mediated vasodilation and the particular susceptibility of young smokers to prolonged exposure to nicotine products. Further careful evaluation of the dose-related effects of nicotine on platelet, leukocyte, hemostatic, and endothelial function and subsequent vascular injury, thrombogenesis, and atherogenesis is indicated.
REFERENCES
- Abdel-Rahman SZ, El-Zein RA. 2000. The 399Gln polymorphism in the DNA repair gene XRCC1 modulates the genotoxic response induced in human lymphocytes by the tobacco-specific nitrosamine NNK. Cancer Lett 159(1):63–71. [PubMed: 10974407]
- Adams MR, Jessup W, Celermajer DS. 1997. Cigarette smoking is associated with increased human monocyte adhesion to endothelial cells: reversibility with oral L-arginine but not vitamin C. J Am Coll Cardiol 29 (491). [PubMed: 9060883]
- Allred EN, Bleecker ER, Chaitman BR, et al. 1989. Short-term effects of carbon monoxide exposure on the exercise performance of subjects with coronary artery disease. N Engl J Med 321:1426. [PubMed: 2682242]
- Anthonisen NR, Connett JE, Kiley JP, et al. 1994. Effects of smoking intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline of FEV1. The Lung Health Study. JAMA 272(19):1497–1505. [PubMed: 7966841]
- Arinami T, Ohtsuki T, Yamakawa-Kobayashi K, et al. 1999. A synergistic effect of serotonin transporter gene polymorphism and smoking in association with CHD. Thromb Haemost 81(6):853–856. [PubMed: 10404755]
- Aronow WS. 1981. Aggravation of angina pectoris by two percent carboxyhemoglobin. Am Heart J 101:154. [PubMed: 7468415]
- Aronow WS, Stemmer EA, Zweig S. 1979. Carbon monoxide and ventricular fibrillation threshold in normal dogs. Arch Environ Health 34:184. [PubMed: 453927]
- Audoly LP, Rocca B, Fabre JE, et al. 2000. Cardiovascular responses to the isoprostanes iPF(2alpha)-III and iPE(2)-III are mediated via the thromboxane A(2) receptor in vivo. Circulation 101(24):2833–2840. [PubMed: 10859290]
- Benedetti A, Comporti M, Esterbauer H. 1980. Identification of 4-hydroxynonenal as a cytotoxic product originating from the peroxidation of liver microsomal lipids. Biochim Biophys Acta 620(2):281–296. [PubMed: 6254573]
- Benowitz NL, Fitzgerald GA, Wilson M, Zhang Q. 1993. Nicotine effects on eicosanoid formation and hemostatic function: comparison of transdermal nicotine and cigarette smoking. J Am Coll Cardiol 22(4):1159–1167. [PubMed: 7691912]
- Blake KV, Gurrin LC, Evans SF, et al. 2000. Maternal cigarette smoking during pregnancy, low birth weight and subsequent blood pressure in early childhood. Early Hum Dev 57(2):137–147. [PubMed: 10735460]
- Bostrom L, Linder LE, Bergstrom J. 1999. Smoking and cervicular fluid levels of IL-6 and TNF-alpha in periodontal disease. J Clin Periodontol 26(6):352–357. [PubMed: 10382574]
- Brash AR, Jackson EK, Saggese CA, Lawson JA, Oates JA, FitzGerald GA. 1983. Metabolic disposition of prostacyclin in humans. J Pharmacol Exp Ther 226(1):78–87. [PubMed: 6345756]
- Brown WV. 2000. Cholesterol lowering in atherosclerosis. American Journal of Cardiology 86(4B):29H–32H. [PubMed: 11021253]
- Burns DM. 2000. Cigarette smoking among the elderly: disease consequences and the benefits of cessation. Am J Health Promot 14(6):357–361. [PubMed: 11067570]
- Campisi R, Czernin J, Schoder H, et al. 1998. Effects of long-term smoking on myocardial blood flow, coronary vasomotion, and vasodilator capacity. Circulation 98(119). [PubMed: 9679717]
- Campisi R, Czernin J, Schoder H, et al. 1999. L-Arginine normalizes coronary vasomotion in long-term smokers. Circulation 99(491). [PubMed: 9927394]
- Chien KL, Lee YT, Sung FC, Su TC, Hsu HC, Lin RS. 1999. Lipoprotein (a) level in the population in Taiwan: relationship to sociodemographic and atherosclerotic risk factors. Atherosclerosis 143(2):267–273. [PubMed: 10217355]
- Cook DG, Shaper AG, Pollock SJ, et al. 1986. Giving up smoking and the risk of heart attacks. Lancet 2(1376). [PubMed: 2878236]
- Craig WY, Paolmaki GE, Haddow JE. 1989. Cigarette smoking and serum lipid and lipoprotein concentrations: an analysis of published data. BMJ 298(784). [PMC free article: PMC1836079] [PubMed: 2496857]
- Cryer PE, Haymond MW, Santiago JV, et al. 1976. Norepinephrine and epinephrine release and adrenergic mediation of smoking-associated hemodynamic and metabolic events. New England Journal of Medicine 295:573. [PubMed: 950972]
- Czernin J, Sun K, Brunken R, et al. 1995. Effect of acute and long-term smoking on myocardial blood flow and flow reserve. Circulation 91:2891. [PubMed: 7796497]
- Davi G, Ciabattoni G, Consoli A, et al. 1999. In vivo formation of 8-iso-prostaglandin f2alpha and platelet activation in diabetes mellitus: effects of improved metabolic control and vitamin E supplementation. Circulation 99(2):224–229. [PubMed: 9892587]
- Davies MJ, Thomas A. 1984. Thrombosis and acute coronary artery lesions in sudden cardiac ischemic death. New England Journal of Medicine 310:1137. [PubMed: 6709008]
- Dawber TR. 1980. The Framingham Study: The Epidemiology of Atherosclerotic Disease . Chapter 8. Cambridge, MA: Harvard University Press.
- DeMatt MP, Pietersma A, Kofflard M, et al. 1996. Association of plasma fibrinogen levels with coronary artery disease, smoking and inflammatory markers. Atherosclerosis 121:185. [PubMed: 9125293]
- Donati MB, Zito F, Castelnuovo AD, Iacoviello L. 2000. Genes, coagulation and cardiovascular risk. J Hum Hypertens 14(6):369–372. [PubMed: 10878696]
- Duthie GG, Arthus JR, Beattie JA, et al. 1993. Cigarette smoking antioxidants, lipid peroxidation, and coronary heart disease. Ann N Y Acad Sci 686(120). [PubMed: 8512243]
- Eliasson B, Attvall S, Taskinen MR, Smith U. 1997. Smoking cessation improves insulin sensitivity in healthy middle-aged men. Eur J Clin Invest 27(5):450–456. [PubMed: 9179554]
- Fitzgerald DJ, Catella F, Roy L, FitzGerald GA. 1988. Marked platelet activation in vivo after intravenous streptokinase in patients with acute myocardial infarction. Circula tion 77(1):142–150. [PubMed: 3275505]
- FitzGerald GA, Oates JA, Novak J. 1988. Cigarette smoking and hemostatic function. Ameri can Heart Journal 115(267). [PubMed: 3276116]
- FitzGerald GA, Pedersen AK, Patrono C. 1983. Analysis of prostacyclin and thromboxane biosynthesis in cardiovascular disease. Circulation 67(6):1174–1177. [PubMed: 6342834]
- FitzGerald GA, Smith B, Pedersen AK, Brash AR. 1984. Increased prostacyclin biosynthesis in patients with severe atherosclerosis and platelet activation. N Engl J Med 310(17):1065–1068. [PubMed: 6231483]
- Fuster V, Chesebro JH, Frye RL, Elveback LR. 1981. Platelet survival and the development of coronary artery disease in the young adult: effects of cigarette smoking, strong family history and medical therapy. Circulation 63(3):546–551. [PubMed: 7460239]
- Gerhardt U, Vorneweg P, Riedasch M, Hohage H. 1999. Acute and persistent effects of smoking on the baroreceptor function. J Auton Pharmacol 19(2):105–108. [PubMed: 10466943]
- Glisic S, Savic I, Alavantic D. 1995. Apolipoprotein B gene DNA polymorphisms (EcoRI and MspI) and serum lipid levels in the Serbian healthy population: interaction of rare alleles and smoking and cholesterol levels. Genet Epidemiol 12(5):499–508. [PubMed: 8557182]
- Gori GB, Lynch CJ. 1985. Analytical cigarette yields as predictors of smoke bioavailability. Regul Toxicol Pharmacol 5(3):314–326. [PubMed: 4059592]
- Green F, Hamsten A, Blomback M, Humphries S. 1993. The role of beta-fibrinogen genotype in determining plasma fibrinogen levels in young survivors of myocardial infarction and healthy controls from Sweden. Thromb Haemost 70(6):915–920. [PubMed: 8165611]
- Griffin BA, Freeman DJ, Tait GW, et al. 1994. Role of plasma triglyceride in the regulation of plasma low density lipoprotein (LDL) subfractions: relative contribution of small, dense LDL to coronary heart disease risk. Atherosclerosis 106(2):241–253. [PubMed: 8060384]
- Hee J, Callais F, Momas I, Laurent AM, Min S, Molinier P, Chastagnier M, Claude JR, Festy B. 1995. Smokers' behaviour and exposure according to cigarette yield and smoking experience. Pharmacology, Biochemistry and Behavior 52(1):195–203. [PubMed: 7501665]
- Heinecke JW. 1999. Mass spectrometric quantification of amino acid oxidation products in proteins: insights into pathways that promote LDL oxidation in the human artery wall. FASEB J 13(10):1113–1120. [PubMed: 10385603]
- Heitzer T, Just H, Munzel T. 1996. a. Antioxidant vitamin C improves endothelial dysfunction in chronic smokers. Circulation 94(6). [PubMed: 8964118]
- Heitzer T, Yia-Herttuala S, Luoma J, et al. 1996. b. Cigarette smoking potentiates endothelial dysfunction of forearm resistance vessels in patients with hypercholesterolemia. Role of oxidized LDL. Circulation 93(1346). [PubMed: 8641023]
- Holm J, Hillarp A, Zoller B, Erhardt L, Berntorp E, Dahlback B. 1999. Factor V Q506 (resistance to activated protein C) and prognosis after acute coronary syndrome. Thromb Haemost 81(6):857–860. [PubMed: 10404756]
- Humphries SE, Henry JA, Montgomery HE. 1999. Gene-environment interaction in the determination of levels of haemostatic variables involved in thrombosis and fibrinolysis. Blood Coagul Fibrinolysis 10 (Suppl 1):S17–21. [PubMed: 10070812]
- Intravenous streptokinase given within 0–4 hours of onset of myocardial infarction reduced mortality in ISIS-2. 1987. Lancet 1(8531):502. [PubMed: 2881054]
- Istvan JA, Lee WW, Buist AS, Connett JE. 1999. Relation of salivary cotinine to blood pressure in middle-aged cigarette smokers. Am Heart J 137(5):928–931. [PubMed: 10220643]
- Kamboh MI, McGarvey ST, Aston CE, Ferrell RE, Bausserman L. 2000. Plasma lipoprotein(a) distribution and its correlates among Samoans. Hum Biol 72(2):321–336. [PubMed: 10803663]
- Kannel WB, McGee DL, Castelli WP. 1984. Latest perspectives on cigarette smoking and cardiovascular disease. The Framingham Study. Journal of Cardiac Rehabilitation 4:267.
- Kawachi I, Colditz GA, Speizer FE, et al. 1997. a. A prospective study of passive smoking and coronary heart disease. Circulation 95(2374). [PubMed: 9170399]
- Kawachi I, Colditz GA, Stampfer MJ, Willett WC, Manson JE, Rosner B, Speizer FE, Hennekens CH. 1994. Smoking cessation and time course of decreased risks of coronary heart disease in middle-aged women. Arch Intern Med 154(2):169–175. [PubMed: 8285812]
- Kawachi I, Colditz GA, Stampfer MJ, Willett WC, Manson JE, Rosner B, Speizer FE, Hennekens CH. 1993. Smoking cessation and decreased risk of stroke in women. JAMA 269(2):232–236. [PubMed: 8417241]
- Kawachi I, Colditz GA, Stampfer MJ, et al. 1997. b. Smoking cessation and decreased risk of total mortality, stroke, and coronary heart disease incidence in women; a prospective cohort study. National Cancer Institute. Changes in Cigarette-Related Disease Risks and Their Implication for Prevention and Control NCI Smoking and Tobacco Control Monograph 8 . Washington, DC: NCI. Pp.531–565.
- Koenig W, Sund M, Frohlich M, et al. 1999. C-Reactive protein, a sensitive marker of inflammation, predicts future risk of coronary heart disease in initially healthy middle-aged men: results from the MONICA (Monitoring Trends and Determinants in Cardiovascular Disease) Augsburg Cohort Study, 1984 to 1992. Circulation 99(2):237–242. [PubMed: 9892589]
- Kugiyama K, Yasue H, Ohgushi M, et. al. 1996. Deficiency in nitric oxide bioactivity in epicardial coronary arteries of cigarette smokers. J Am Coll Cardiol 8(1161). [PubMed: 8890810]
- Kunapuli P, Lawson JA, Rokach JA, Meinkoth JL, FitzGerald GA. 1998. Prostaglandin F2alpha (PGF2alpha) and the isoprostane, 8, 12-iso-isoprostane F2alpha-III, induce cardiomyocyte hypertrophy. Differential activation of downstream signaling pathways. J Biol Chem 273(35):22442–22452. [PubMed: 9712868]
- Lassila R, Seyberth HW, Haapanen A, Schweer H, Koskenvuo M, Laustiola KE. 1988. Vaso-active and atherogenic effects of cigarette smoking: a study of monozygotic twins discordant for smoking. BMJ 297(6654):955–957. [PMC free article: PMC1834683] [PubMed: 3142565]
- Lawson JA, Rokach J, FitzGerald GA. 1999. Isoprostanes: formation, analysis and use as indices of lipid peroxidation in vivo. J Biol Chem 274(35):24441–24444. [PubMed: 10455102]
- Lewis HD, Davis JW, Archibald DG, et al. 1983. Protective effects of aspirin against acute myocardial infarction and death in men with unstable angina. Results of a Veterans Administration Cooperative Study. N Engl J Med 309(7):396–403. [PubMed: 6135989]
- Li R, Boerwinkle E, Olshan AF, et al. 2000. Glutathione S-transferase genotype as a susceptibility factor in smoking-related coronary heart disease. Atherosclerosis 149(2):451–462. [PubMed: 10729397]
- McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. 1999. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci U S A 96(1):272–277. [PMC free article: PMC15129] [PubMed: 9874808]
- McAdam BF, Mardini IA, Habib A, et al. 2000. Effect of regulated expression of human cyclooxygenase isoforms on eicosanoid and isoeicosanoid production in inflammation. J Clin Invest 105(10):1473–1482. [PMC free article: PMC315469] [PubMed: 10811855]
- McNamara P, Lawson JA, Rokach J, FitzGerald GA. 1999. F2 and E2 isoprostanes are ligands for peroxisome prolifer-activated receptors. FASEB J 13:A1549.
- McGinnis JM, Foege WH. 1999. Mortality and morbidity attributable to use of addictive substances in the United States. Proceedings of the Association of American Physicians 111(2):109–118. [PubMed: 10220805]
- Meade TW, Vickers MV, Thompson SG, Stirling Y, Haines AP, Miller GJ. 1985. Epidemiological characteristics of platelet aggregability. Br Med J (Clin Res Ed) 290(6466):428–432. [PMC free article: PMC1417797] [PubMed: 3918615]
- Mikhailidis DP, Papadakis JA, Ganotakis ES. 1998. Smoking, diabetes and hyperlipidaemia. J R Soc Health 118(2):91–93. [PubMed: 10076642]
- Mikkelsen KL, Winberg N, Hoegholm A, et al. 1997. Smoking related to 24-h ambulatory blood pressure and heart rate: a study in 352 normotensive Danish subjects. Am J Hypertens 10(5 Pt 1):483–491. [PubMed: 9160757]
- Minami J, Ishimitsu T, Matsuoka H. 1999. Effects of smoking cessation on blood pressure and heart rate variability in habitual smokers. Hypertension 33(1 Pt 2):586–590. [PubMed: 9931170]
- Moauad J, Fernandez F, Herbert JL, et al. 1986. Cigarette smoking during coronary angiography: diffuse or focal narrowing (spasm) of the coronary arteries in 13 patients with angina at rest and normal coronary angiograms. Cathet Cardiovasc Diagn 12:366. [PubMed: 3102072]
- Moliterno DJ, Willard JE, Lange RA, et al. 1994. Coronary-artery vasoconstriction induced by cocaine, cigarette smoking, or both. New England Journal of Medicine 330:454. [PubMed: 8289850]
- Mormando RM. 2000. Lipid levels. Applying the second National Cholesterol Educaton Program report to geriatric medicine. Geriatrics 55(8):48–53. [PubMed: 10953686]
- Morrow JD, Frei B, Longmire AW, et al. 1995. Increase in circulating products of lipid peroxidation (F2-isoprostanes) in smokers. Smoking as a cause of oxidative damage. N Engl J Med 332(18):1198–1203. [PubMed: 7700313]
- Mundal R, Kjeldsen SE, Sandvik L, Erikssen G, Thaulow E, Erikssen J. 1997. Predictors of 7-year changes in exercise blood pressure: effects of smoking, physical fitness and pulmonary function. J Hypertens 15(3):245–249. [PubMed: 9468451]
- Murata T, Ushikubi F, Matsuoka T, et al. 1997. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature 388(6643):678–682. [PubMed: 9262402]
- Nardo CJ, Chambless LE, Light KC, et al. 1999. Descriptive epidemiology of blood pressure response to change in body position. The ARIC study. Hypertension 33(5):1123–1129. [PubMed: 10334798]
- Newby DE, Wright RA, Labinjoh C, et al. 1999. Endothelial dysfunction, impaired endogenous fibrinolysis, and cigarette smoking: a mechanism for arterial thrombosis and myocardial infarction. Circulation 99:1411. [PubMed: 10086962]
- Nicholson NS, Smith SL, Fuller GC. 1984. Effect of the stable endoperoxide analog U-46619 on prostacyclin production and cyclic AMP levels in bovine endothelial cells. Thromb Res 35(2):183–192. [PubMed: 6089372]
- Nicod P, Rehr R, Winniford MD, et al. 1984. Acute systemic and coronary hemodynamic and serologic responses to cigarette smoking in long-term smokers with atherosclerotic coronary artery disease. J Am Coll Cardiol 4:964. [PubMed: 6548482]
- Nowak J, Murray JJ, Oates JA, FitzGerald GA. 1987. Biochemical evidence of a chronic abnormality in platelet and vascular function in healthy individuals who smoke cigarettes. Circulation 76(1):6–14. [PubMed: 3297389]
- Nygard O, Refsum H, Ueland PM, Vollset SE. 1998. Major lifestyle determinants of plasma total homocysteine distribution: the Hordaland Homocysteine Study. Am J Clin Nutr 67(2):263–270. [PubMed: 9459374]
- Nyboe J, Jensen G, Appleyard M, Schnohr P. 1991. Smoking and the risk of first acute myocardial infarction. American Heart Journal 122:438–447. [PubMed: 1858623]
- O'Malley PG, Taylor AJ, Jackson JL, Doherty TM, Detrano RC. 2000. Prognostic value of coronary electron-beam computed tomography for coronary heart disease events in asymptomatic populations. Am J Cardiol 85(8):945–948. [PubMed: 10760331]
- Oates JA. 1989. Aspirin in the prevention of catastrophes of the coronary circulation. Samuelsson B, editor. , ed. Advances in Prostaglandin, Thromboxane and Leukotriene Research . New York: Raven. Pp. 57. Chapter 8. [PubMed: 2526539]
- Okasha M, McCarron P, McEwen J, Davey Smith G. 2000. Determinants of adolescent blood pressure: findings from the Glasgow University student cohort. J Hum Hypertens 14(2):117–124. [PubMed: 10723118]
- PDAY (Pathobiological Determinants of Atherosclerosis in Youth Research Group). 1990. The pathobiological determinants of atherosclerosis in Youth Research Group: relationship of atherosclerosis in young men to serum lipoprotein cholesterol concentrations and smoking. JAMA 265:3018. [PubMed: 2243430]
- Penn A, Snyder CA. 1996. 1,3 Butadiene, a vapor phase component of environmental tobacco smoke, accelerates arteriosclerotic plaque development. Circulation 93:552. [PubMed: 8565175]
- Pennathur S, Jackson-Lewis V, Przedborski S, Heinecke JW. 1999. Mass spectrometric quantification of 3-nitrotyrosine, ortho-tyrosine, and o,o′-dityrosine in brain tissue of 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine-treated mice, a model of oxidative stress in Parkinson's disease. J Biol Chem 274(49):34621–34628. [PubMed: 10574926]
- Pratico D, Tangirala RK, Rader DJ, Rokach J, FitzGerald GA. 1998. Vitamin E suppresses isoprostane generation in vivo and reduces atherosclerosis in ApoE-deficient mice. Nat Med 4(10):1189–1192. [PubMed: 9771755]
- Pryor WA. 1997. Cigarette smoke radicals and the role of free radicals in chemical carcinogenicity. Environ Health Perspect 105 Suppl 4(1):875–882. [PMC free article: PMC1470037] [PubMed: 9255574]
- Quillen JE, Rossen JD, Oskarsson JJ, et al. 1993. Acute effect of cigarette smoking on the coronary circulation: constriction of epicardial and resistance vessels. J Am Coll Cardiol 22:642. [PubMed: 8354792]
- Raggi P, Callister TQ, Cooil B, et al. 2000. Identification of patients at increased risk of first unheralded acute myocardial infarction by electron-beam computed tomography. Cir culation 101(8):850–855. [PubMed: 10694523]
- Raitakari OT, Adams MR, McCredie RJ, Griffiths KA, Stocker R, Celermajer DS. 2000. Oral vitamin C and endothelial function in smokers: short-term improvement, but no sustained beneficial effect. J Am Coll Cardiol 35(6):1616–1621. [PubMed: 10807468]
- Reed DM, MacLean CJ, Hayashi T. 1987. Predictors of atherosclerosis in the Honolulu Heart Program: I. Biologic, dietary and lifestyle characteristics. American Journal of Epidemiol ogy 126:214. [PubMed: 3605050]
- Reilly M, Delanty N, Lawson JA, FitzGerald GA. 1996. Modulation of oxidant stress in vivo in chronic cigarette smokers. Circulation 94(1):19–25. [PubMed: 8964113]
- Reinders JH, Brinkman HJ, van Mourik JA, de Groot PG. 1986. Cigarette smoke impairs endothelial cell prostacyclin production. Arteriosclerosis 6(1):15–23. [PubMed: 3080000]
- Reis RP, Azinheira J, Reis HP, Pina JE, Correia JM, Luis AS. 2000. Influence of smoking on homocysteinemia at baseline and after methionine load. Rev Port Cardiol 19(4):471–474. [PubMed: 10874843]
- Ridker PM, Rifai N, Stampfer MJ, Hennekens CH. 2000. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circula tion 101(15):1767–1772. [PubMed: 10769275]
- Roberts LJ, Morrow JD. 2000. Measurement of F(2)-isoprostanes as an index of oxidative stress in vivo. Free Radic Biol Med 28(4):505–513. [PubMed: 10719231]
- Rosenberg L, Kaufman DW, Helrich SP, et. al. 1985. The risk of myocardial infarction after quitting smoking in men under 55 years of age. New England Journal of Medicine 313:1511. [PubMed: 4069159]
- Rosenberg L, Palmer JR, Shapiro S. 1990. Declining risk of myocardial infarction among women who stopped smoking. New England Journal of Medicine 322:213. [PubMed: 2294448]
- Saw SM. 1999. Homocysteine and atherosclerotic disease: the epidemiologic evidence. Ann Acad Med Singapore 28(4):565–568. [PubMed: 10561772]
- Seman LJ, DeLuca C, Jenner JL, et al. 1999. Lipoprotein(a)-cholesterol and coronary heart disease in the Framingham Heart Study. Clin Chem 45(7):1039–1046. [PubMed: 10388480]
- Sen-Banerjee S, Siles X, Campos H. 2000. Tobacco smoking modifies association between Gln-Arg192 polymorphism of human paraoxonase gene and risk of myocardial infarction. Arterioscler Thromb Vasc Biol 20(9):2120–2126. [PubMed: 10978258]
- Shinagawa K, Tokimoto T, Shirane K. 1998. Spin trapping of nitric oxide in aqueous solutions of cigarette smoke. Biochem Biophys Res Commun 253(1):99–103. [PubMed: 9875226]
- Shinozaki K, Hattori Y, Suzuki M, et al. 1997. Insulin resistance as an independent risk factor for carotid artery wall intima media thickening in vasospastic angina. Arterioscler Thromb Vasc Biol 17(11):3302–3310. [PubMed: 9409326]
- Sinzinger H, Granegger S, Oguogho A. 2000. Passive smoking in children is associated with increased isoprostane 8-EPI-PGF. Atherosclerosis 151:257.
- Steenland K, Thun M, Lally C, Heath C. 1996. Environmental tobacco smoke and coronary heart disease in the American Cancer Society CPS-II cohort. Circulation 94(4):622–628. [PubMed: 8772680]
- Stellman SD, Garfinkel L. 1986. Smoking habits and tar levels in a new American Cancer Society prospective study of 1.2 million men and women. Journal of the National Cancer Institute 76(6):1057–1063. [PubMed: 3458944]
- Tahtinen TM, Vanhala MJ, Oikarinen JA, Keinanen-Kiukaanniemi SM. 1998. Effect of smoking on the prevalence of insulin resistance-associated cardiovascular risk factors among Finnish men in military service. J Cardiovasc Risk 5(5–6):319–323. [PubMed: 9920003]
- Takizawa H, Tanaka M, Takami K, et al. 2000. Increased expression of inflammatory mediators in small-airway epithelium from tobacco smokers. Am J Physiol Lung Cell Mol Physiol 278(5):L906–913. [PubMed: 10781420]
- Targher G, Alberiche M, Zenere MB, Bonadonna RC, Muggeo M, Bonora E. 1997. Cigarette smoking and insulin resistance in patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab 82(11):3619–3624. [PubMed: 9360516]
- Taylor AE, Johnson DC, Kazemi H. 1992. Environmental tobacco smoke and cardiovascular disease. A position paper from the Council on Cardiopulmonary and critical Care, American Heart Association. Circulation 86(2):699–702. [PubMed: 1638735]
- The Pooling Project Research Group. 1978. Relationship of blood pressure, serum cholesterol, smoking habit, relative weight and ECG abnormalities to incidence of major coronary events: final report of the pooling project. Journal of Chronic Disease 31(201). [PubMed: 681498]
- The sixth report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. 1997. Archives of Internal Medicine 157(21):2413–2446. [PubMed: 9385294]
- Thom SR, Xu YA, Ischiropoulos H. 1997. Vascular endothelial cells generate peroxynirite in response to carbon monoxide exposure. Chem Res Toxicol 10:1023. [PubMed: 9305585]
- Thun MJ, Myers DG, Day-Lally C, et al. 1997. Age and exposure-response relationships between cigarette smoking and premature death in cancer prevention study II. National Cancer Institute. Changes in Cigarette-Related Disease Risks and Their Implication for Prevention and Control. NCI Smoking and Tobacco Control Monograph 8 . Washington, DC: NCI. Pp.383–475.
- U.S. DHHS (U.S. Department of Health and Human Services). 1989. The Health Consequences of Smoking: 25 Years of Progress. A Report of the Surgeon General . Chapter 8. Washington, DC: DHHS Publications.
- U.S. DHHS (U.S. Department of Health and Human Services). 1983. The Health Consequences of Smoking: Cardiovascular Disease . Chapter 8. Washington, DC: DHHS Publications.
- Valkonen MM, Kuusi T. 2000. Vitamin C prevents the acute atherogenic effects of passive smoking. Free Radic Biol Med 28(3):428–436. [PubMed: 10699755]
- Vita JA, Treasure CB, Yeung AC, et al. 1992. Patients with evidence of coronary endothelial dysfunction as assessed by acetylcholine infusion demonstrate marked increase in sensitivity to constrictor effects of catecholamines. Circulation 85:1390. [PubMed: 1555281]
- Weber C, Erl W, Weber K, et al. 1996. Increased adhesiveness of isolated monocytes to endothelium is prevented by vitamin C intake in smokers. Circulation 93(1488). [PubMed: 8608614]
- Wennmalm A, Benthin G, Granstrom EF, Persson L, Petersson AS, Winell S. 1991. Relation between tobacco use and urinary excretion of thromboxane A2 and prostacyclin metabolites in young men. Circulation 83(5):1698–1704. [PubMed: 2022025]
- Winniford MD, Wheelan KR, Kremers MS, et al. 1986. Smoking-induced coronary vasoconstriction in patients with atherosclerotic coronary artery disease: evidence for adrenergically mediated alterations in coronary artery tone. Circulation 73:662. [PubMed: 3948369]
- Yamaguchi Y, Kagota S, Haginaka J, Kunitomo M. 2000. Peroxynitrite-generating species: good candidate oxidants in aqueous extracts of cigarette smoke. Jpn J Pharmacol 82(1):78–81. [PubMed: 10874594]
- Zeiher AM, Drexler H, Wollschlaeger H, et al. 1989. Coronary vasomotion in response to sympathetic stimulation in humans: importance of the functional integrity of the endothelium. J Am Coll Cardiol 14:1181. [PubMed: 2808971]
- Zeiher AM, Drexler H, Wollschlager H, et al. 1991. Endothelial dysfunction of the coronary microvasculature is associated with coronary blood flow regulation in patients with early atherosclerosis. Circulation 84(1984). [PubMed: 1934373]
- Cardiovascular Disease - Clearing the SmokeCardiovascular Disease - Clearing the Smoke
- Spinal Muscle Atrophy - StatPearlsSpinal Muscle Atrophy - StatPearls
- Microbe sample from candidate division CPR1 bacterium ADurb.Bin160Microbe sample from candidate division CPR1 bacterium ADurb.Bin160biosample
- BioSample links for Nucleotide (Select 1167320610) (1)BioSample
Your browsing activity is empty.
Activity recording is turned off.
See more...