

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
McEntyre J, Ostell J, editors. The NCBI Handbook [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2002-.

This publication is provided for historical reference only and the information may be out of date.

Summary
The GenBank sequence database is an annotated collection of all publicly available nucleotide sequences and their protein translations. This database is produced at National Center for Biotechnology Information (NCBI) as part of an international collaboration with the European Molecular Biology Laboratory (EMBL) Data Library from the European Bioinformatics Institute (EBI) and the DNA Data Bank of Japan (DDBJ). GenBank and its collaborators receive sequences produced in laboratories throughout the world from more than 100,000 distinct organisms. GenBank continues to grow at an exponential rate, doubling every 10 months. Release 134, produced in February 2003, contained over 29.3 billion nucleotide bases in more than 23.0 million sequences. GenBank is built by direct submissions from individual laboratories, as well as from bulk submissions from large-scale sequencing centers.
Direct submissions are made to GenBank using BankIt, which is a Web-based form, or the stand-alone submission program, Sequin. Upon receipt of a sequence submission, the GenBank staff assigns an Accession number to the sequence and performs quality assurance checks. The submissions are then released to the public database, where the entries are retrievable by Entrez or downloadable by FTP. Bulk submissions of Expressed Sequence Tag (EST), Sequence Tagged Site (STS), Genome Survey Sequence (GSS), and High-Throughput Genome Sequence (HTGS) data are most often submitted by large-scale sequencing centers. The GenBank direct submissions group also processes complete microbial genome sequences.
History
Initially, GenBank was built and maintained at Los Alamos National Laboratory (LANL). In the early 1990s, this responsibility was awarded to NCBI through congressional mandate. NCBI undertook the task of scanning the literature for sequences and manually typing the sequences into the database. Staff then added annotation to these records, based upon information in the published article. Scanning sequences from the literature and placing them into GenBank is now a rare occurrence. Nearly all of the sequences are now deposited directly by the labs that generate the sequences. This is attributable to, in part, a requirement by most journal publishers that nucleotide sequences are first deposited into publicly available databases (DDBJ/EMBL/GenBank) so that the Accession number can be cited and the sequence can be retrieved when the article is published. NCBI began accepting direct submissions to GenBank in 1993 and received data from LANL until 1996. Currently, NCBI receives and processes about 20,000 direct submission sequences per month, in addition to the approximately 200,000 bulk submissions that are processed automatically.
International Collaboration
In the mid-1990s, the GenBank database became part of the International Nucleotide Sequence Database Collaboration with the EMBL database (European Bioinformatics Institute, Hinxton, United Kingdom) and the Genome Sequence Database (GSDB; LANL, Los Alamos, NM). Subsequently, the GSDB was removed from the Collaboration (by the National Center for Genome Resources, Santa Fe, NM), and DDBJ (Mishima, Japan) joined the group. Each database has its own set of submission and retrieval tools, but the three databases exchange data daily so that all three databases should contain the same set of sequences. Members of the DDBJ, EMBL, and GenBank staff meet annually to discuss technical issues, and an international advisory board meets with the database staff to provide additional guidance. An entry can only be updated by the database that initially prepared it to avoid conflicting data at the three sites.
The Collaboration created a Feature Table Definition that outlines legal features and syntax for the DDBJ, EMBL, and GenBank feature tables. The purpose of this document is to standardize annotation across the databases. The presentation and format of the data are different in the three databases, however, the underlying biological information is the same.
Confidentiality of Data
When scientists submit data to GenBank, they have the opportunity to keep their data confidential for a specified period of time. This helps to allay concerns that the availability of their data in GenBank before publication may compromise their work. When the article containing the citation of the sequence or its Accession number is published, the sequence record is released. The database staff request that submitters notify GenBank of the date of publication so that the sequence can be released without delay. The request to release should be sent to vog.hin.mln.ibcn@nimda-bg.
Direct Submissions
The typical GenBank submission consists of a single, contiguous stretch of DNA or RNA sequence with annotations. The annotations are meant to provide an adequate representation of the biological information in the record. The GenBank Feature Table Definition describes the various features and subsequent qualifiers agreed upon by the International Nucleotide Sequence Database Collaboration.
Currently, only nucleotide sequences are accepted for direct submission to GenBank. These include mRNA sequences with coding regions, fragments of genomic DNA with a single gene or multiple genes, and ribosomal RNA gene clusters. If part of the nucleotide sequence encodes a protein, a conceptual translation, called a CDS (coding sequence), is annotated. The span of the CDS feature is mapped to the nucleotide sequence encoding the protein. A protein Accession number (/protein_id) is assigned to the translation product, which will subsequently be added to the protein databases.
Multiple sequences can be submitted together. Such batch submissions of non-related sequences may be processed together but will be displayed in Entrez (Chapter 15) as single records. Alternatively, by using the Sequin submission tool (Chapter 12), a submitter can specify that several sequences are biologically related. Such sequences are classified as environmental sample sets, population sets, phylogenetic sets, mutation sets, or segmented sets. Each sequence within a set is assigned its own Accession number and can be viewed independently in Entrez. However, with the exception of segmented sets, each set is also indexed within the PopSet division of Entrez, thus allowing scientists to view the relationship between the sequences.
What defines a set? Environmental sample, population, phylogenetic, and mutation sets all contain a group of sequences that spans the same gene or region of the genome. Environmental samples are derived from a group of unclassified or unknown organisms. A population set contains sequences from different isolates of the same organism. A phylogenetic set contains sequences from different organisms that are used to determine the phylogenetic relationship between them. Sequencing multiple mutations within a single gene gives rise to a mutation set.
All sets, except segmented sets, may contain an alignment of the sequences within them and might include external sequences already present in the database. In fact, the submitter can begin with an existing alignment to create a submission to the database using the Sequin submission tool. Currently, Sequin accepts FASTA+GAP, PHYLIP, MACAW, NEXUS Interleaved, and NEXUS Contiguous alignments. Submitted alignments will be displayed in the PopSet section of Entrez.
Segmented sets are a collection of noncontiguous sequences that cover a specified genetic region. The most common example is a set of genomic sequences containing exons from a single gene where part or all of the intervening regions have not been sequenced. Each member record within the set contains the appropriate annotation, exon features in this case. However, the mRNA and CDS will be annotated as joined features across the individual records. Segmented sets themselves can be part of an environmental sample, population, phylogenetic, or mutation set.
Bulk Submissions: High-Throughput Genomic Sequence (HTGS)
HTGS entries are submitted in bulk by genome centers, processed by an automated system, and then released to GenBank. Currently, about 30 genome centers are submitting data for a number of organisms, including human, mouse, rat, rice, and Plasmodium falciparum, the malaria parasite.
HTGS data are submitted in four phases of completion: 0, 1, 2, and 3. Phase 0 sequences are one-to-few reads of a single clone and are not usually assembled into contigs. They are low-quality sequences that are often used to check whether another center is already sequencing a particular clone. Phase 1 entries are assembled into contigs that are separated by sequence gaps, the relative order and orientation of which are not known (Figure 1). Phase 2 entries are also unfinished sequences that may or may not contain sequence gaps. If there are gaps, then the contigs are in the correct order and orientation. Phase 3 sequences are of finished quality and have no gaps. For each organism, the group overseeing the sequencing effort determines the definition of finished quality.
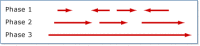
Figure
Figure 1. Diagram showing the orientation and gaps that might be expected in high-throughput sequence from phases 1, 2, and 3.
Phase 0, 1, and 2 records are in the HTG division of GenBank, whereas phase 3 entries go into the taxonomic division of the organism, for example, PRI (primate) for human. An entry keeps its Accession number as it progresses from one phase to another but receives a new Accession.Version number and a new gi number each time there is a sequence change.
Submitting Data to the HTG Division
To submit sequences in bulk to the HTG processing system, a center or group must set up an FTP account by writing to vog.hin.mln.ibcn@nimda-sgth. Submitters frequently use two tools to create HTG submissions, Sequin or fa2htgs. Both of these tools require FASTA-formatted sequence, i.e., a definition line beginning with a "greater than" sign (">") followed by a unique identifier for the sequence. The raw sequence appears on the lines after the definition line. For sequences composed of contigs separated by gaps, a modified FASTA format is used. In addition, Sequin users must modify the Sequin configuration file so that the HTG genome center features are enabled.
fa2htgs is a command-line program that is downloaded to the user's computer. The submitter invokes a script with a series of parameters (arguments) to create a submission. It has an advantage over Sequin in that it can be set up by the user to create submissions in bulk from multiple files.
Submissions to HTG must contain three identifiers that are used to track each HTG record: the genome center tag, the sequence name, and the Accession number. The genome center tag is assigned by NCBI and is generally the FTP account login name. The sequence name is a unique identifier that is assigned by the submitter to a particular clone or entry and must be unique within the group's submissions. When a sequence is first submitted, it has only a sequence name and genome center tag; the Accession number is assigned during processing. All updates to that entry must include the center tag, sequence name, and Accession number, or processing will fail.
The HTG Processing Pathway
Submitters deposit HTGS sequences in the form of Seq-submit files generated by Sequin, fa2htgs, or their own ASN.1 dumper tool into the SEQSUBMIT directory of their FTP account. Every morning, scripts automatically pick up the files from the FTP site and copy them to the processing pathway, as well as to an archive. Once processing is complete and if there are no errors in the submission, the files are automatically loaded into GenBank. The processing time is related to the number of submissions that day; therefore, processing can take from one to many hours.
Entries can fail HTG processing because of three types of problems:
- 1.
Formatting: submissions are not in the proper Seq-submit format.
- 2.
Identification: submissions may be missing the genome center tag, sequence name, or Accession number, or this information is incorrect.
- 3.
Data: submissions have problems with the data and therefore fail the validator checks.
When submissions fail HTG processing, a GenBank annotator sends email to the sequencing center, describing the problem and asking the center to submit a corrected entry. Annotators do not fix incorrect submissions; this ensures that the staff of the submitting genome center fixes the problems in their database as well.
The processing pathway also generates reports. For successful submissions, two files are generated: one contains the submission in GenBank flat file format (without the sequence); and another is a status report file. The status report file, ac4htgs, contains the genome center, sequence name, Accession number, phase, create date, and update date for the submission. Submissions that fail processing receive an error file with a short description of the error(s) that prevented processing. The GenBank annotator also sends email to the submitter, explaining the errors in further detail.
Additional Quality Assurance
When successful submissions are loaded into GenBank, they undergo additional validation checks. If GenBank annotators find errors, they write to the submitters, asking them to fix these errors and submit an update.
Whole Genome Shotgun Sequences (WGS)
Genome centers are taking multiple approaches to sequencing complete genomes from a number of organisms. In addition to the traditional clone-based sequencing whose data are being submitted to HTGS, these centers are also using a WGS approach to sequence the genome. The shotgun sequencing reads are assembled into contigs, which are now being accepted for inclusion in GenBank. WGS contig assemblies may be updated as the sequencing project progresses and new assemblies are computed. WGS sequence records may also contain annotation, similar to other GenBank records.
Each sequencing project is assigned a stable project ID, which is made up of four letters. The Accession number for a WGS sequence contains the project ID, a two-digit version number, and six digits for the contig ID. For instance, a project would be assigned an Accession number AAAX00000000. The first assembly version would be AAAX01000000. The last six digits of this ID identify individual contigs. A master record for each assembly is created. This master record contains information that is common among all records of the sequencing project, such as the biological source, submitter, and publication information. There is also a link to the range of Accession numbers for the individual contigs in this assembly.
WGS submissions can be created using tbl12asn, a utility that is packaged with the Sequin submission software. Information on submitting these sequences can be found at Whole Genome Shotgun Submissions.
Bulk Submissions: EST, STS, and GSS
Expressed Sequence Tags (EST), Sequence Tagged Sites (STSs), and Genome Survey Sequences (GSSs) sequences are generally submitted in a batch and are usually part of a large sequencing project devoted to a particular genome. These entries have a streamlined submission process and undergo minimal processing before being loaded to GenBank.
ESTs are generally short (<1 kb), single-pass cDNA sequences from a particular tissue and/or developmental stage. However, they can also be longer sequences that are obtained by differential display or Rapid Amplification of cDNA Ends (RACE) experiments. The common feature of all ESTs is that little is known about them; therefore, they lack feature annotation.
STSs are short genomic landmark sequences (1). They are operationally unique in that they are specifically amplified from the genome by PCR amplification. In addition, they define a specific location on the genome and are, therefore, useful for mapping.
GSSs are also short sequences but are derived from genomic DNA, about which little is known. They include, but are not limited to, single-pass GSSs, BAC ends, exon-trapped genomic sequences, and Alu PCR sequences.
EST, STS, and GSS sequences reside in their respective divisions within GenBank, rather than in the taxonomic division of the organism. The sequences are maintained within GenBank in the dbEST, dbSTS, and dbGSS databases.
Submitting Data to dbEST, dbSTS, or dbGSS
Because of the large numbers of sequences that are submitted at once, dbEST, dbSTS, and dbGSS entries are stored in relational databases where information that is common to all sequences can be shared. Submissions consist of several files containing the common information, plus a file of the sequences themselves. The three types of submissions have different requirements, but all include a Publication file and a Contact file. See the dbEST, dbSTS, and dbGSS pages for the specific requirements for each type of submission.
In general, users generate the appropriate files for the submission type and then email the files to vog.hin.mln.ibcn@bus-hctab. If the files are too big for email, they can be deposited into a FTP account. Upon receipt, the files are examined by a GenBank annotator, who fixes any errors when possible or contacts the submitter to request corrected files. Once the files are satisfactory, they are loaded into the appropriate database and assigned Accession numbers. Additional formatting errors may be detected at this step by the data-loading software, such as double quotes anywhere in the file or invalid characters in the sequences. Again, if the annotator cannot fix the errors, a request for a corrected submission is sent to the user. After all problems are resolved, the entries are loaded into GenBank.
Bulk Submissions: HTC and FLIC
HTC records are High-Throughput cDNA/mRNA submissions that are similar to ESTs but often contain more information. For example, HTC entries often have a systematic gene name (not necessarily an official gene name) that is related to the lab or center that submitted them, and the longest open reading frame is often annotated as a coding region.
FLIC records, Full-Length Insert cDNA, contain the entire sequence of a cloned cDNA/mRNA. Therefore, FLICs are generally longer, and sometimes even full-length, mRNAs. They are usually annotated with genes and coding regions, although these may be lab systematic names rather than functional names.
HTC Submissions
HTC entries are usually generated with Sequin or tbl2asn, and the files are emailed to vog.hin.mln.ibcn@bus-bg. If the files are too big for email, then by prior arrangement, the submitter can deposit the files by FTP and send a notification to vog.hin.mln.ibcn@nimda-bg that files are on the FTP site.
HTC entries undergo the same validation and processing as non-bulk submissions. Once processing is complete, the records are loaded into GenBank and are available in Entrez and other retrieval systems.
FLIC Submissions
FLICs are processed via an automated FLIC processing system that is based on the HTG automated processing system. Submitters use the program tbl2asn to generate their submissions. As with HTG submissions, submissions to the automated FLIC processing system must contain three identifiers: the genome center tag, the sequence name (SeqId), and the Accession number. The genome center tag is assigned by NCBI and is generally the FTP account login name. The sequence name is a unique identifier that is assigned by the submitter to a particular clone or entry and must be unique within the group's FLIC submissions. When a sequence is first submitted, it has only a sequence name and genome center tag; the Accession number is assigned during processing. All updates to that entry include the center tag, sequence name, and Accession number, or processing will fail.
The FLIC Processing Pathway
The FLIC processing system is analogous to the HTG processing system. Submitters deposit their submissions in the FLICSEQSUBMIT directory of their FTP account and notify us that the submissions are there. We then run the scripts to pick up the files from the FTP site and copy them to the processing pathway, as well as to an archive. Once processing is complete and if there are no errors in the submission, the files are automatically loaded into GenBank.
As with HTG submissions, FLIC entries can fail for three reasons: problems with the format, problems with the identification of the record (the genome center, the SeqId, or the Accession number), or problems with the data itself. When submissions fail FLIC processing, a GenBank annotator sends email to the sequencing center, describing the problem and asking the center to submit a corrected entry. Annotators do not fix incorrect submissions; this ensures that the staff of the submitting genome center fixes the problems in their database as well. At the completion of processing, reports are generated and deposited in the submitter's FTP account, as described for HTG submissions.
Submission Tools
Direct submissions to GenBank are prepared using one of two submission tools, BankIt or Sequin.
BankIt
BankIt is a Web-based form that is a convenient and easy way to submit a small number of sequences with minimal annotation to GenBank. To complete the form, a user is prompted to enter submitter information, the nucleotide sequence, biological source information, and features and annotation pertinent to the submission. BankIt has extensive Help documentation to guide the submitter. Included with the Help document is a set of annotation examples that detail the types of information that are required for each type of submission. After the information is entered into the form, BankIt transforms this information into a GenBank flatfile for review. In addition, a number of quality assurance and validation checks ensure that the sequence submitted to GenBank is of the highest quality. The submitter is asked to include spans (sequence coordinates) for the coding regions and other features and to include amino acid sequence for the proteins that derive from these coding regions. The BankIt validator compares the amino acid sequence provided by the submitter with the conceptual translation of the coding region based on the provided spans. If there is a discrepancy, the submitter is requested to fix the problem, and the process is halted until the error is resolved. To prevent the deposit of sequences that contain cloning vector sequence, a BLAST similarity search is performed on the sequence, comparing it to the VecScreen database. If there is a match to this database, the user is asked to remove the contaminating vector sequence from their submission or provide an explanation as to why the screen was positive. Completed forms are saved in ASN.1 format, and the entry is submitted to the GenBank processing queue. The submitter receives confirmation by email, indicating that the submission process was successful.
Sequin
Sequin is more appropriate for complicated submissions containing a significant amount of annotation or many sequences. It is a stand-alone application available on NCBI's FTP site. Sequin creates submissions from nucleotide and amino acid sequences in FASTA format with tagged biological source information in the FASTA definition line. As in BankIt, Sequin has the ability to predict the spans of coding regions. Alternatively, a submitter can specify the spans of their coding regions in a five-column, tab-delimited table and import that table into Sequin. For submitting multiple, related sequences, e.g., those in a phylogenetic or population study, Sequin accepts the output of many popular multiple sequence-alignment packages, including FASTA+GAP, PHYLIP, MACAW, NEXUS Interleaved, and NEXUS Contiguous. It also allows users to annotate features in a single record or a set of records globally. For more information on Sequin, see Chapter 12.
Completed Sequin submissions should be emailed to GenBank at vog.hin.mln.ibcn@bus-bg. Larger files may be submitted by SequinMacrosend.
Sequence Data Flow and Processing: From Laboratory to GenBank
Triage
All direct submissions to GenBank, created either by Sequin or BankIt, are processed by the GenBank annotation staff. The first step in processing submissions is called triage. Within 48 hours of receipt, the database staff reviews the submission to determine whether it meets the minimal criteria for incorporation into GenBank and then assigns an Accession number to each sequence. All sequences must be >50 bp in length and be sequenced by, or on behalf of, the group submitting the sequence. GenBank will not accept sequences constructed in silico; noncontiguous sequences containing internal, unsequenced spacers; or sequences for which there is not a physical counterpart, such as those derived from a mix of genomic DNA and mRNA. Submissions are also checked to determine whether they are new sequences or updates to sequences submitted previously. After receiving Accession numbers, the sequences are put into a queue for more extensive processing and review by the annotation staff.
Indexing
Triaged submissions are subjected to a thorough examination, referred to as the indexing phase. Here, entries are checked for:
- 1.
Biological validity. For example, does the conceptual translation of a coding region match the amino acid sequence provided by the submitter? Annotators also ensure that the source organism name and lineage are present, and that they are represented in NCBI's taxonomy database. If either of these is not true, the submitter is asked to correct the problem. Entries are also subjected to a series of BLAST similarity searches to compare the annotation with existing sequences in GenBank.
- 2.
Vector contamination. Entries are screened against NCBI's UniVec database to detect contaminating cloning vector.
- 3.
Publication status. If there is a published citation, PubMed and MEDLINE identifiers are added to the entry so that the sequence and publication records can be linked in Entrez.
- 4.
Formatting and spelling. If there are problems with the sequence or annotation, the annotator works with the submitter to correct them.
Completed entries are sent to the submitter for a final review before release into the public database. If the submitters requested that their sequences be released after processing, they have 5 days to make changes prior to release. The submitter may also request that GenBank hold their sequence until a future date. The sequence must become publicly available once the Accession number or the sequence has been published. The GenBank annotation staff currently processes about 1,900 submissions per month, corresponding to approximately 20,000 sequences.
GenBank annotation staff must also respond to email inquiries that arrive at the rate of approximately 200 per day. These exchanges address a range of topics including:
- updates to existing GenBank records, such as new annotation or sequence changes
- problem resolution during the indexing phase
- requests for release of the submitter's sequence data or an extension of the hold date
- requests for release of sequences that have been published but are not yet available in GenBank
- lists of Accession numbers that are due to appear in upcoming issues of a publisher's journals
- reports of potential annotation problems with entries in the public database
- requests for information on how to submit data to GenBank
One annotator is responsible for handling all email received in a 24-hour period, and all messages must be acted upon and replied to in a timely fashion. Replies to previous emails are forwarded to the appropriate annotator.
Processing Tools
The annotation staff uses a variety of tools to process and update sequence submissions. Sequence records are edited with Sequin, which allows staff to annotate large sets of records by global editing rather than changing each record individually. This is truly a time saver because more than 100 entries can be edited in a single step (see Chapter 12 on Sequin for more details). Records are stored in a database that is accessed through a queue management tool that automates some of the processing steps, such as looking up taxonomy and PubMed data, starting BLAST jobs, and running automatic validation checks. Hence, when an annotator is ready to start working on an entry, all of this information is ready to view. In addition, all of the correspondence between GenBank staff and the submitter is stored with the entry. For updates to entries already present in the public database, the live version of the entry is retrieved from ID, and after making changes, the annotator loads the entry back into the public database. This entry is available to the public immediately after loading.
Microbial Genomes
The GenBank direct submissions group has processed more than 50 complete microbial genomes since 1996. These genomes are relatively small in size compared with their eukaryotic counterparts, ranging from five hundred thousand to five million bases. Nonetheless, these genomes can contain thousands of genes, coding regions, and structural RNAs; therefore, processing and presenting them correctly is a challenge. Currently, the DDBJ/EMBL/GenBank Nucleotide Sequence Database Collaboration has a 350-kilobase (kb) upper size limit for sequence entries. Because a complete bacterial genome is larger than this arbitrary limit, it must be split into pieces. GenBank routinely splits complete microbial genomes into 10-kb pieces with a 60-bp overlap between pieces. Each piece contains approximately 10 genes. A CON entry, containing instructions on how to put the pieces back together, is also made. The CON entry contains descriptor information, such as source organism and references, as well as a join statement providing explicit instructions on how to generate the complete genome from the pieces. The Accession number assigned to the CON record is also added as a secondary Accession number on each of the pieces that make up the complete genome (see Figure 2).

Figure
Figure 2. The information toward the bottom of the record describes how to generate the complete genome from the pieces.
Submitting and Processing Data
Submitters of complete genomes are encouraged to contact us at vog.hin.mln.ibcn@semoneg before preparing their entries. A FTP account is required to submit large files, and the submission should be deposited at least 1 month before publication to allow for processing time and coordinated release before publication. In addition, submitters are required to follow certain guidelines, such as providing unique identifiers for proteins and systematic names for all genes. Entries should be prepared with the submission tool tbl2asn, a utility that is part of the Sequin package (Chapter 12). This utility creates an ASN.1 submission file from a five-column, tab-delimited file containing feature annotation, a FASTA-formatted nucleotide sequence, and an optional FASTA-formatted protein sequence.
Complete genome submissions are reviewed by a member of the GenBank annotation staff to ensure that the annotation and gene and protein identifiers are correct, and that the entry is in proper GenBank format. Any problems with the entry are resolved through communication with the submitter. Once the record is complete, the genome is carefully split into its component pieces. The genome is split so that none of the breaks occurs within a gene or coding region. A member of the annotation staff performs quality assurance checks on the set of genome pieces to ensure that they are correct and representative of the complete genome. The pieces are then loaded into GenBank, and the CON record is created.
The microbial genome records in GenBank are the building blocks for the Microbial Genome Resources in Entrez Genomes.
Third Party Annotation (TPA) Sequence Database
The vast amount of publicly available data from the human genome project and other genome sequencing efforts is a valuable resource for scientists throughout the world. A laboratory studying a particular gene or gene family may have sequenced numerous cDNAs but has neither the resources nor inclination to sequence large genomic regions containing the genes, especially when the sequence is available in public databases. The researcher might choose then to download genomic sequences from GenBank and perform experimental analyses on these sequences. However, because this researcher did not perform the sequencing, the sequence, with its new annotations, cannot be submitted to DDBJ/EMBL/GenBank. This is unfortunate because important scientific information is being excluded from the public databases. To address this problem, the International Nucleotide Sequence Database Collaboration established a separate section of the database for such TPA (see Third Party Annotation Sequence Database).
All sequences in the TPA database are derived from the publicly available collection of sequences in DDBJ/EMBL/GenBank. Researchers can submit both new and alternative annotations of genomic sequence to GenBank. TPA entries can be also created by combining the exon sequences from genomic sequences or by making contigs of EST sequences to make mRNA sequences. TPA submissions must use sequence data that are already represented in DDBJ/EMBL/GenBank, have annotation that is experimentally supported, and appear in a peer-reviewed scientific journal. TPA sequences will be released to the public database only when their Accession numbers and/or sequence data appear in a peer-reviewed publication in a biological journal.
Appendix: GenBank, RefSeq, TPA and UniProt: What’s in a Name?
The National Center for Biotechnology Information (NCBI) often is asked about the differences between its GenBank, RefSeq, and TPA databases and how they relate to the UniProt database. This document was prepared in response to those inquiries, and more specifically to a request from attendees at a 2006 workshop on microbial genomes held at NCBI and attended by bacterial annotation groups, sequencing centers, and members of the American Society for Microbiology (ASM). The article originally was published in the May 2007 issue of the American Society for Microbiology’s journal Microbe (http://www.microbemagazine.org/index.php?option=com_content&view=article&id=1270:genbank-refseq-tpa-and-uniprot-whats-in-a-name&catid=347:letters&Itemid=419). While there was some input from the European Bioinformatics Institute on UniProt and Swiss-Prot for the document, it represents an NCBI perspective.
GenBank
NCBI’s GenBank database is a collection of publicly available annotated nucleotide sequences, including mRNA sequences with coding regions, segments of genomic DNA with a single gene or multiple genes, and ribosomal RNA gene clusters.
GenBank is specifically intended to be an archive of primary sequence data. Thus, to be included, the sequencing must have been conducted by the submitter. NCBI does some quality control checks and will notify a submitter if something appears amiss, but it does not curate the data; the author has the final say on the sequence and annotation placed in the GenBank record. Authors are encouraged to update their records with new sequence or annotation data, but in practice records are seldom updated.
Records can be updated only by the author, or by a third party if the author has given them permission and notified NCBI. This delegation of authority has happened in a limited number of cases, generally where a genome sequence was determined by a lab or sequencing center and updating rights were subsequently given to a model organism database, which then took over ongoing maintenance of annotation.
Because GenBank is an archival database and includes all sequence data submitted, there are multiple entries for some loci. Just as the primary literature includes similar experiments conducted under slightly different conditions, GenBank may include many sequencing results for the same loci. These different sequencing submissions can reflect genetic variations between individuals or organisms, and analyzing these differences is one way of identifying single nucleotide polymorphisms.
GenBank exchanges data daily with its two partners in the International Nucleotide Sequence Database Collaboration (INSDC): the European Bioinformatics Institute (EBI) of the European Molecular Biology Laboratory (EMBL), and the DNA Data Bank of Japan (DDBJ). Nearly all sequence data are deposited into INSDC databases by the labs that generate the sequences, in part because journal publishers generally require deposition prior to publication so that an accession number can be included in the paper.
If part of a GenBank nucleotide sequence encodes a protein, a conceptual translation – called a coding region or coding sequence (CDS) – is annotated. A protein accession number (a "protein id") is assigned to the translation product and is noted on the GenBank record. This protein id is linked to a record for the protein sequence in NCBI’s protein databases. In the UniProt database, described later, these sequences are contained in the TrEMBL (Translated EMBL) portion of the database.
See the GenBank overview at http://www.ncbi.nlm.nih.gov/Genbank/index.html.
RefSeq
The Reference Sequence (RefSeq) database is a curated collection of DNA, RNA, and protein sequences built by NCBI. Unlike GenBank, RefSeq provides only one example of each natural biological molecule for major organisms ranging from viruses to bacteria to eukaryotes. For each model organism, RefSeq aims to provide separate and linked records for the genomic DNA, the gene transcripts, and the proteins arising from those transcripts. RefSeq is limited to major organisms for which sufficient data is available (almost 4,000 distinct “named” organisms as of January 2007), while GenBank includes sequences for any organism submitted (approximately 250,000 different named organisms).
To produce RefSeq records, NCBI culls the best available information on each molecule and updates the records as more information emerges. A commonly used analogy is that if GenBank is akin to the primary research literature, RefSeq is akin to the review literature.
In some cases, creation of a RefSeq record involves no more than selecting a single good example from GenBank and making a copy in RefSeq, which credits the GenBank record. In other cases, NCBI in-house staff generates and annotates the records based on the existing primary data, sometimes by combining parts of several GenBank records. Also, some records are automatically imported from other curated databases, such as the SGD database of yeast genome data and the FlyBase database of Drosophila genomes (for a list of RefSeq collaborators see www.ncbi.nlm.nih.gov/RefSeq/collaborators). The approach selected for creating a RefSeq record depends on the specific organism and the quality of information available.
When NCBI first creates a RefSeq record, the record initially reflects only the information from the source GenBank record with added links. At this point, the record has not yet been reviewed by NCBI staff, and therefore it is identified as “provisional.” After NCBI examines the record – often adding information from other GenBank records, such as the sequences for the 5’UTR and 3’UTR, and providing further literature references – it is marked as “reviewed.”
RefSeq records appear in a similar format as the GenBank records from which they are derived. However, they can be distinguished from GenBank records by their accession prefix, which includes an underscore, and a notation in the “comment” field that indicates the RefSeq status. RefSeq records can be accessed through NCBI’s Nucleotide and Protein databases, which are among the many databases linked through the Entrez search and retrieval system. When retrieving search results, users can choose to see all GenBank records or only RefSeq records by clicking on the appropriate tab at the top of the results page. Users also can choose to search only RefSeq records, or specific types of RefSeq records (such as mRNAs), by using the “Limits” feature in Entrez. Further information about the database can be obtained at the RefSeq homepage.
Key Characteristics of GenBank versus RefSeq
| GenBank | RefSeq |
|---|---|
| Not curated | Curated |
| Author submits | NCBI creates from existing data |
| Only author can revise | NCBI revises as new data emerge |
| Multiple records for same loci common | Single records for each molecule of major organisms |
| Records can contradict each other | |
| No limit to species included | Limited to model organisms |
| Data exchanged among INSDC members | Exclusive NCBI database |
| Akin to primary literature | Akin to review articles |
| Proteins identified and linked | Proteins and transcripts identified and linked |
| Access via NCBI Nucleotide databases | Access via Nucleotide & Protein databases |
TPA
The Third Party Annotation (TPA) database contains sequences that are derived or assembled from sequences already in the INSDC databases. Whereas DDBJ, EMBL and GenBank contain primary sequence data and corresponding annotations submitted by the laboratories that did the sequencing, the TPA database contains nucleotide sequences built from the existing primary data with new annotation that has been published in a peer-reviewed scientific journal. The database includes two types of records: experimental (supported by wet-lab evidence) and inferential (where the annotation is inferred and not the subject of direct experimentation).
TPA bridges the gap between GenBank and RefSeq, permitting authors publishing new experimental evidence to re-annotate sequences in a public database as they think best, even if they were not the primary sequencer or the curator of a model organism database. These records are part of the INSDC collaboration, and thus appear in all three databases (GenBank, DDBJ and EMBL).
Like GenBank and RefSeq records, TPA records can be retrieved through the Nucleotide section of Entrez. The TPA records can be distinguished from other records by the definition line, which begins with the letters "TPA," and by the Keywords field, which states "Third Party Annotation; TPA." Users can restrict their search to TPA data by selecting the database in the Properties search field or by adding the command “AND tpa[prop]” to their query. The database is significantly smaller than GenBank, with about one record for every 12,000 in GenBank. Details about how to submit data and examples of what can and cannot be submitted to TPA are provided on the TPA homepage.
UniProt
UniProt (Universal Protein Resource) is a protein sequence database that was formed through the merger of three separate protein databases: the Swiss Institute of Bioinformatics’ and the European Bioinformatics Institute’s Swiss-Prot and TrEMBL (Translated EMBL Nucleotide Sequence Data Library) databases, and Georgetown University’s PIR-PSD (Protein Information Resource Protein Sequence Database).
Swiss-Prot and TrEMBL continue as two separate sections of the UniProt database. The Swiss-Prot component consists of manually annotated protein sequence records that have added information, such as binding sites for drugs. The TrEMBL portion consists of computationally analyzed sequence records that are awaiting full manual annotation; following curation, they are transferred to Swiss-Prot.
TrEMBL is derived from the CDS translations annotated on records in the INSDC databases, with some additional computational merging and adjustment. Given the very high rate of sequencing, and the effort it takes to do manual annotation, the Swiss-Prot component of UniProt is generally much smaller than the TrEMBL component. Because Swiss-Prot’s manual annotation provides much additional information, NCBI’s protein databases provide links to Swiss-Prot records, even if the sequence is the same as one or more INSDC translations.
Key Characteristics of UniProt versus GenBank and RefSeq
| UniProt | GenBank and RefSeq |
|---|---|
| Produced by SIB, EBI & Georgetown U. | Produced by INSDC and NCBI |
| Protein data only | Protein and nucleotide data |
| Curated in Swiss-Prot, not in TrEMBL | Curated in RefSeq, not in GenBank |
References
- 1.
- Olson M, Hood L, Cantor C, Botstein D. A common language for physical mapping of the human genome. Science. 1989;245(4925):1434–1435. [PubMed: 2781285]
- Summary
- History
- International Collaboration
- Confidentiality of Data
- Direct Submissions
- Bulk Submissions: High-Throughput Genomic Sequence (HTGS)
- Whole Genome Shotgun Sequences (WGS)
- Bulk Submissions: EST, STS, and GSS
- Bulk Submissions: HTC and FLIC
- Submission Tools
- Sequence Data Flow and Processing: From Laboratory to GenBank
- Microbial Genomes
- Third Party Annotation (TPA) Sequence Database
- Appendix: GenBank, RefSeq, TPA and UniProt: What’s in a Name?
- References
- GenBank: The Nucleotide Sequence Database - The NCBI HandbookGenBank: The Nucleotide Sequence Database - The NCBI Handbook
Your browsing activity is empty.
Activity recording is turned off.
See more...