Clinical Description
Age at diagnosis. Brugada syndrome manifests primarily during adulthood, with a mean age of sudden death of approximately 40 years. The youngest individual was diagnosed at two days of life and the oldest was diagnosed at age 85 years [Huang & Marcus 2004].
Sex differences. Although Brugada syndrome is more prevalent among males, it affects females as well, and both sexes are at a high risk for ventricular arrhythmias and sudden death [Hong et al 2004b].
Presentation. Currently, the most common presentation is that of a person in the fifth decade with malignant arrhythmias and a previous history of syncopal episodes. Syncope is a common presenting symptom [Mills et al 2005, Benito & Brugada 2006, Karaca & Dinckal 2006].
Affected individuals in whom sustained ventricular arrhythmias are easily induced and who have a spontaneously abnormal EKG have a 45% likelihood of having an arrhythmic event at any time during life [Benito et al 2009]. Electrical storms (also known as arrhythmic storms) – multiple episodes of ventricular arrhythmias that occur over a short period of time – are malignant but rare phenomena in Brugada syndrome. Incessant ventricular tachycardia (VT) is defined as hemodynamically stable VT continuing for hours.
Brugada syndrome can occur in conjunction with conduction disease. The presence of first-degree AV block, intraventricular conduction delay, right bundle branch block, and sick sinus syndrome in Brugada syndrome is not unusual [Smits et al 2005].
Clinical presentations of Brugada syndrome may also include sudden infant death syndrome (SIDS; death of a child during the first year of life without an identifiable cause) [Priori et al 2000, Antzelevitch 2001, Skinner et al 2005, Van Norstrand et al 2007] and sudden unexpected nocturnal death syndrome (SUNDS) [Vatta et al 2002], a syndrome seen in Southeast Asia in which young people die from cardiac arrest with no identifiable cause. The same pathogenic variant in SCN5A was identified in individuals with Brugada syndrome and SUNDS, thus supporting the hypothesis that they are the same disease [Hong et al 2004a].
Precipitating factors for the Brugada EKG pattern and the syndrome of sudden cardiac death include fever, cocaine use, electrolyte disturbances, and use of class I antiarrhythmic medications and a number of other noncardiac medications [Francis & Antzelevitch 2005]. Most importantly, in some (usually young) persons, the presence of the induced EKG pattern has been associated with sudden cardiac death. The pathophysiologic mechanisms behind this association remain largely unknown.
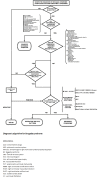
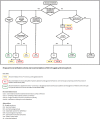
Predicting risk of malignant arrhythmias. Several parameters have been investigated to improve stratification of the risk of developing malignant arrhythmias (see ).
Proposed risk stratification scheme and recommendations of ICD in individuals with Brugada syndrome Reproduced from Berne & Brugada [2012] with permission
Inducibility during electrophysiologic study (EPS) is the only parameter currently used for clinical decision making. During such a study the heart is electrically stimulated using intracardiac catheters. Although the inducibility of arrhythmias in an asymptomatic individual during the EPS is highly predictive of subsequent malignant events (arrhythmias and sudden cardiac death), the data remain controversial. Several groups do not use EPS for risk stratification in asymptomatic individuals. Several multiparametric approaches to determine risk are available. However, their predictive abilities remain modest in individuals with Brugada syndrome and in asymptomatic individuals [
Rodríguez-Mañero et al 2022]. Thus, decisions regarding timing of implantation of a defibrillator vary widely among physicians and investigators [
Eckardt et al 2005,
Glatter et al 2005,
Ikeda et al 2005,
Al-Khatib 2006,
Delise et al 2006,
Gehi et al 2006,
Imaki et al 2006,
Ito et al 2006,
Ott & Marcus 2006,
Tatsumi et al 2006,
Benito et al 2009].
Genotype has been proposed as an additional parameter for risk stratification.
Meregalli et al [2009] found that among individuals with an
SCN5A pathogenic variant, those who were more symptomatic had more EKG signs of conduction slowing, supporting the notion that conduction slowing, mediated by
loss-of-function SCN5A pathogenic variants, was a key pathophysiologic mechanism in Brugada syndrome. This limited study indicates that it may be possible in the future to use
genotype information in risk stratification; however, at present this remains an area of investigation.
Pathophysiology. Brugada syndrome, caused by a sodium channelopathy, is associated with age-related progressive conduction abnormalities, such as prolongation of the EKG PQ, QRS, and HV intervals [Smits et al 2002, Yokokawa et al 2007]. Sodium current dysfunction contributes to local conduction block in the epicardium, resulting in multiple spikes within the QRS complex and triggering of atrial and ventricular fibrillation [Morita et al 2008].
Sodium channelopathies exhibited typical Brugada-type EKG and frequent arrhythmogenesis during bradycardia [Makiyama et al 2005]; both quinidine and isoproterenol normalized the J-ST elevation and prevented arrhythmias.
Nomenclature
Vatta et al [2002] and Hong et al [2004a] determined that sudden unexpected nocturnal death syndrome (SUNDS) and Brugada syndrome are phenotypically, genetically, and functionally the same disorder. SUNDS was originally described in individuals from Southeast Asia. Other names for SUNDS include sudden and unexpected death syndrome (SUDS), bangungut (Philippines), non-lai tai (Laos), lai-tai (Thailand), and pokkuri (Japan).
Prevalence
Brugada syndrome occurs worldwide. The prevalence of the disease in endemic areas (South Asia) is on the order of 1:2,000 persons. In countries in Southeast Asia in which SUNDS is endemic, it is the second leading cause of death (following accidents) of men under age 40 years.
Data from published studies indicate that Brugada syndrome is responsible for 4%-12% of unexpected sudden deaths and for up to 20% of all sudden death in individuals with an apparently normal heart [Brugada et al 1999a].
A prospective study of an adult Japanese population (22,027 individuals) showed 12 individuals (prevalence of 0.05%) with EKGs compatible with Brugada syndrome [Tohyou et al 1995]. A second study of adults in Awa, Japan, showed a prevalence of 0.6% (66:10,420 individuals) [Namiki et al 1995]. In contrast, a third study in Japanese children showed only a 0.0006% (1:163,110) prevalence of EKGs compatible with Brugada syndrome [Hata et al 1997]. Therefore, in the absence of symptoms and/or molecular genetic testing, these studies provide an estimate of the prevalence of the Brugada syndrome EKG pattern (not of Brugada syndrome) in the population studied. The results suggest that Brugada syndrome manifests primarily during adulthood, a finding in concordance with the mean age of sudden death (age 35-40 years).