Clinical Description
Primary phenotypic features associated with EZH2-related overgrowth include tall stature, macrocephaly, and intellectual disability, which are observed in association with a characteristic facial appearance (round face, flattened occiput, hypertelorism, almond-shaped palpebral fissures, retrognathia, large, fleshy ears, and a "stuck on" appearance to the chin) [Tatton-Brown et al 2013, Imagawa et al 2023].
The phenotypic spectrum associated with germline EZH2 pathogenic variants is broad, with classic EZH2-related Weaver syndrome at one end of the spectrum and tall stature at the other. Although most individuals diagnosed with a heterozygous germline EZH2 pathogenic variant have been identified because of a clinical suspicion of Weaver syndrome, a minority have been identified through molecular genetic testing of family members of probands or individuals with overgrowth who did not have a clinical diagnosis of Weaver syndrome [Tatton-Brown et al 2011, Gibson et al 2012]. Thus, the full extent of the phenotypic spectrum of heterozygous EZH2 pathogenic variants is not yet known.
To date, at least 68 individuals have been identified with a pathogenic variant in EZH2 [Tatton-Brown et al 2011, Gibson et al 2012, Al-Salem et al 2013, Tatton-Brown et al 2013, Cohen et al 2016, Usemann et al 2016, Lui et al 2018, Griffiths et al 2019, Turkkahraman et al 2021, Imagawa et al 2023, Oh et al 2023]. The following description of the phenotypic features associated with this condition is based on these reports.
Growth. From data available on 23 newborns, the mean birth length was 2.2 standard deviations (SD) above the mean, with a range of 0.5 SD below the mean to 4.9 SD above the mean. The mean birth weight of 45 newborns was 1.7 SD above the mean, with a range of 1.6 SD below the mean to 4.6 SD above the mean [Tatton-Brown et al 2013].
Tall stature is a near-consistent finding: height in 59 of 65 individuals was at least two SD above the mean (ages 1-70 years). Of note, three of four individuals with a height less than two SD above the mean had been tall as young children. The mean postnatal height was 3.5 SD above the mean.
Of 59 individuals for whom information is available, 27 had a head circumference less than two SD above the mean and 32 had macrocephaly (>2 SD above the mean), with a head circumference reaching up to 5.5 SD above the mean. Head circumferences at birth were at least two SD above the mean in 5 of 6 infants for whom data is available [Gibson et al 2012, Cohen et al 2016]. These data suggest that macrocephaly in EZH2-related overgrowth is likely to be present from birth but longitudinal follow up is required.
Neurologic. Ventriculomegaly, reported in seven individuals, was generally associated with normal cerebrospinal fluid pressure and did not require shunting [Gibson et al 2012, Tatton-Brown et al 2013, Griffiths et al 2019]. Other reported brain MRI findings include neuronal migration defects (including polymicrogyria in several individuals), periventricular leukomalacia (two individuals), and cerebellar abnormalities (two individuals).
Intellectual disability in those with a brain MRI abnormality was:
Mild in seven individuals (ventriculomegaly [5 individuals], periventricular leukomalacia [1 individual], and cerebellar hypoplasia [1 individual]);
Moderate in three individuals (periventricular leukomalacia with ventriculomegaly [1 individual] and isolated ventriculomegaly [2 individuals]);
Severe in two individuals with polymicrogyria and pachygyria [
Tatton-Brown et al 2013,
Griffiths et al 2019]; in contrast, the individual with polymicrogyria reported by
Al-Salem et al [2013] had normal developmental milestones and body asymmetry (left side smaller than the right) with brisk reflexes and increased tone on the left. The degree of intellectual disability of the individual with polymicrogyria reported by
Cohen et al [2016] is unknown.
Note: The degree of intellectual disability was not reported for one individual with ventriculomegaly.
Several reported individuals with EZH2-related overgrowth had seizures. Four individuals had afebrile seizures [Gibson et al 2012, Usemann et al 2016, Griffiths et al 2019]. Seizure types include tonic-clonic (age of onset 13 years) [1 individual] and brief absence (age of onset 15 years) [Gibson et al 2012]. Two individuals had seizures associated with a febrile illness [Tatton-Brown et al 2013, Cohen et al 2016].
Abnormal tone. In general, abnormal tone (hypotonia, hypertonia, or mixed central hypotonia and peripheral hypertonia), if present, resolves during childhood.
Hypotonia (predominantly central) was reported in 22 of 47 individuals.
Hypertonia (predominantly peripheral manifesting as stiffness in the limbs with brisk reflexes) was reported in 14 of 51 individuals.
Note: Three of the individuals presenting with peripheral hypertonia were also reported to have central hypotonia [
Tatton-Brown et al 2013].
Cognitive features. Information on cognitive function is available for 61 individuals. Eight had normal intellect; 52 individuals had variable intellectual disability (ID), including the following:
Mild ID (30/61). Children attend mainstream school but need some extra help – e.g., a statement of educational needs – and are expected to live independently as adults and likely to have their own family.
Moderate ID (13/61). Children develop speech and need a high level of support in mainstream education but are likely to require special educational needs. While unlikely to live independently as adults, they may live in sheltered accommodation or with additional support.
Severe ID (3/61). Individuals require special education during school and are likely to require considerable support during adulthood.
Unclassified ID (6/61). Insufficient information was provided regarding degree of ID.
Behavioral issues including autistic features, phobias, and anxiety have been anecdotally reported [Tatton-Brown et al 2013].
Skeletal features
Advanced bone age. Of 33 individuals evaluated, all had advanced bone age.
Scoliosis was reported in 13 individuals and pectus abnormalities (excavatum or carinatum) in four individuals. Scoliosis ranged from severe (early-childhood onset requiring surgical intervention) to mild (requiring monitoring but no therapeutic intervention).
Camptodactyly. Some affected individuals had camptodactyly of the fingers, some had camptodactyly of the toes, and some had camptodactyly of fingers and toes. On occasion, the toe camptodactyly required surgical correction.
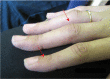
Adult boutonniere deformity. Several adults developed hyperextension of the distal interphalangeal joints and flexion of the proximal interphalangeal joints of the hands analogous to a mild boutonniere deformity (see ).
Talipes equinovarus. Eight individuals had talipes equinovarus ranging from fixed and bilateral (requiring surgery) in two individuals to mild (unilateral that resolved with physiotherapy) in three.
Mild hyperextension of the distal interphalangeal joints and flexion of the proximal interphalangeal joints in a woman age 22 years with a heterozygous EZH2 pathogenic variant
Connective tissue features
Ligamentous laxity. While ligamentous laxity with associated joint hypermobility and pes planus is common, it is not usually reported unless complicated by joint pain. Individuals with EZH2-related overgrowth are frequently reported to have poor coordination that may be (at least partially) attributable to lax ligaments.
Skin that was soft and doughy to the touch has been reported in 23 of 43 affected children.
Umbilical hernia has been seen in 28 of 57 children and was sufficiently large to require surgery in eight neonates.
Poor feeding has been reported in 10 of 28 neonates, including one who required nasogastric tube feeding for two weeks. Although poor feeding may be attributable to neonatal hypotonia, this was only reported in three of the infants with poor feeding.
Hoarse, low-pitched cry was reported in 16 of 35 affected infants.
Tumors have been reported in seven of 68 affected individuals [Tatton-Brown et al 2013, Usemann et al 2016, Cohen et al 2016, Oh et al 2023]. A summary of reported tumor types is provided in Table 3.
Table 3.
Tumor Types Reported in Individuals with EZH2-Related Overgrowth
View in own window
| Tumor Type | # of Persons | Age | EZH2 Pathogenic Variant |
|---|
| Neuroblastoma | 5 | 13 months | c.2044G>A (p.Ala682Thr) |
| 4 years | c.458A>G (p.Tyr153Cys) |
| Neonate | c.394C>T (p.Pro132Ser) |
| Neonate | c.398A>G (p.Tyr133Cys) |
| 7 months | c.2050C>T (p.Arg684Cys) |
| Pre-T-cell non-Hodgkin lymphoma | 1 | 13 years | c.2233G>A (p.Glu745Lys) |
| Acute lymphoblastic leukemia | 1 | 13 months | c.2044G>A (p.Ala682Thr) |
| Acute myeloid leukemia | 1 | 16 years | c.395C>T (p.Pro132Leu) |
Additional clinical features reported in a small number of individuals (and therefore possibly not associated with the EZH2 pathogenic variant) are included for completeness:
Café au lait macules (2 individuals), hemangioma (4 individuals), pigmented nevi (3 individuals)
Large hands and feet (1 individual)
Hypermetropia (hyperopia; 3 individuals), strabismus (3 individuals), myopia (1 individual)
Hydrocele (2 individuals), cryptorchidism (1 individual), hypospadias (1 individual)
Cleft palate (3 individuals)
Hearing loss (conductive and sensorineural; 3 individuals)
Cardiac anomalies (4 individuals), including mitral valve prolapse (1 individual), ventricular septal defect (2 individuals), and patent ductus arteriosus (1 individual)
Gastroesophageal reflux (1 individual), hiatal hernia (1 individual)
Neonatal hypoglycemia (2 individuals)
Neonatal hypocalcemia (2 individual)