Basic pluripotent stem cell culture protocols*
Contributors
Authors:
Maria Borowski,
1,* Maria Giovino-Doherty,
2 Lan Ji,
3 Meng-Jiao Shi,
4 Kelly P Smith,
5 and
Joseph Laning6.

Affiliations
1 Massachusetts Stem Cell Bank, University of Massachusetts Medical School, Shrewsbury, MA 01545 USA
2 Massachusetts Stem Cell Bank, University of Massachusetts Medical School, Shrewsbury, MA 01545 USA
3 Massachusetts Stem Cell Bank, University of Massachusetts Medical School, Shrewsbury, MA 01545 USA
4 Massachusetts Stem Cell Bank, University of Massachusetts Medical School, Shrewsbury, MA 01545 USA
5 Massachusetts Stem Cell Bank, University of Massachusetts Medical School, Shrewsbury, MA 01545 USA
6 Massachusetts Stem Cell Bank, University of Massachusetts Medical School, Shrewsbury, MA 01545 USA
To whom correspondence should be addressed. E-mail: maria.borowski@umassmed.edu
Published June 10, 2012.
Stem cell research is a rapidly expanding field with the potential to develop therapeutic agents to treat diseases as well as study disease development from early stages. The culture of human pluripotent stem cells shares many of the same protocols as standard mammalian cell culture. However, the successful culture and maintenance of human pluripotent stem cells (hPSCs) in an undifferentiated state requires additional considerations to ensure that cells maintain their key characteristics of self-renewal and pluripotency. There are several basic techniques needed for the culturing of mammalian cells, including thawing frozen stocks, plating cells in culture vessels, changing media, passaging and cryopreservation. The protocols in this document represent a subset of the standard operating procedures used to maintain and culture stem cells at the Massachusetts Human Stem Cell Bank, and have been thoroughly testing and verified.
A Stem cell culture considerations
Stem cell research is a rapidly expanding field with the potential to develop therapeutic agents to treat diseases as well as study disease development from early stages. However, to fulfill this promise, researchers need to have access to standardized protocols for the development, maintenance and differentiation of these unique cells. Such “best practices” will allow comparisons of different studies and hasten the refinement of these techniques. Such standardization can be driven by resources such as StemBook and by stem cell banks. In addition to standardization of these best practices, stem cell banks also serve as valuable resources of properly identified, quality controlled and characterized cell lines as well as helping to navigate the legal and IP issues that are common to working with stem cells.
The protocols in this document represent a subset of the standard operating procedures used to maintain and culture stem cells at the Massachusetts Human Stem Cell Bank, and have been thoroughly tested and verified.
A1 Successful stem cell culture
The culture of human pluripotent stem cells shares many of the same protocols as standard mammalian cell culture. However, the successful culture and maintenance of human pluripotent stem cells (hPSCs) in an undifferentiated state requires additional considerations to ensure that cells maintain their key characteristics of self-renewal and pluripotency.
Successful hPSC culture requires the recreation of the in vivo stem cell microenvironment, or “niche”, which includes growth factors, cell-to-cell interactions and cell to matrix adhesions. Unlike many cell types, hPSCs are grown in aggregates, or colonies, which helps create this niche. Standard culture of hPSCs involves exposure to media enriched with growth factors found in fetal bovine serum (FBS) or defined serum replacements. In addition, standard hPSC culture systems utilize support cells such as an inactivated mouse embryonic fibroblast (MEF) feeder layer to support growth and prevent differentiation. These cells provide necessary intercellular interactions, extracellular scaffolding and factors creating a robust and stable hPSC culture environment.
There are several basic techniques needed for the culturing of mammalian cells, including thawing frozen stocks, plating cells in culture vessels, changing media, passaging and cryopreservation. Passaging refers to the removal of cells from their current culture vessel and transferring them to one or more new culture vessels. Passaging is necessary to reduce the harmful effects of overcrowding and for expansion of the culture. Protocols provided in this manual describe the standard culture of hPSCs. These procedures have been shown to be reliable and generate reproducible experimental data.
Although the standard culture protocols described here use a variety of animal products, any clinical use of hPSCs will require elimination of these products as they pose a risk of exposure to retroviruses and other pathogens from the culture environment. Many approaches have been published for culturing hPSCs in an entirely animal-free environment, including the use of human fibroblasts and serum, the use of defined substrates, and replacement of serum with defined growth factors.1-3
A2 Quality control of cell cultures
It is essential that researchers ensure the sterility, authenticity and genetic stability of cell lines used in their work in order to publish and provide reproducible and informative experimental data. Upon receipt of a new cell line, it is highly recommended that cells assayed for the criteria outlined below and monitored at regular intervals to confirm these characteristics.
Cell line sterility
Cells in continuous culture are generally vulnerable to microbial contamination. Bacterial and fungal contamination can cause cell death and eventual loss of entire cultures. Human stem cell cultures are particularly susceptible since they are commonly cultured in enriched media without antibiotics. It is highly recommended that cells received from any provider should be tested for microbial contamination before initial use and at regular intervals during routine culture in the laboratory. Since microbes are everywhere in the culture environment, stringent practice of aseptic technique is essential for preventing culture contamination.
In addition to bacteria and fungi, another common contaminant is mycoplasma. These intracellular microorganisms are generally smaller than bacteria and may affect cell growth, particularly at high levels of contamination. However, persistent infections of cells can result in genetic and phenotypic changes. Common sources of mycoplasma include contaminated materials of animal origin such as serum, trypsin and primary feeder cell cultures. Thus it is important to test for the presence of mycoplasma on a regular basis and discard contaminated cultures. Common methods used to detect mycoplasma include enzymatic assays, polymerase chain reaction (PCR), culture in selective media, and DNA staining of test cells to visualize mycoplasma that grow in close association with the cell membrane. Although the least sensitive, DNA staining with 4′6-Diamidino-2-phenylindole (DAPI), a fluorescent stain that binds strongly to DNA, is a simple method that can be employed in most laboratories to detect mycoplasma. Commercially available kits are offered by a number of reagent companies.
Viruses are another form of contamination. Viruses can alter a cell line's characteristics to varying degrees through multiple activities. These include utilization of host cellular resources for viral replication and integration into the host genome. Viral infection may affect experimental data and could result in misleading interpretations. In addition, cultures that are contaminated with blood borne viruses capable of human infection pose a serious health risk. Common sources of viral contamination include animal products and preparations such as bovine serum, antibodies and mouse embryonic fibroblasts.6,7 Although testing for viral contamination is conducted by stem cell banks and repositories on a regular basis, this testing is not common practice for individual research laboratories. It is recommended that laboratories test cells for viral infection prior to their distribution and that recipient laboratories request documentation of this testing. Several companies offer a wide range of viral testing services; their fees depend on the breadth and types of tests performed.
Cell line authenticity
Cells in culture are at high risk for cross-contamination since most laboratories routinely culture multiple cell lines simultaneously. Studies have shown that up to 30% of cell lines donated to public repositories are contaminated by rapidly growing cell types7,8 as was the case with HeLa cells, which can outgrow and replace the original cell lines. This cross-contamination can be more subtle and lead to false data and misleading conclusions. This is of particular interest in regulated environments where authenticity or identity is a key component of quality controls systems for clinical applications.
Authentication of cell lines obtained from outside sources prior to their use in experiments is essential and can be achieved by comparing the unique features of received cell lines against those of the original isolate. Several approaches used to authenticate cell lines. Genotyping takes advantage of the small genetic variations between individual cell lines. Current DNA typing employs PCR-based techniques to analyze similar hypervariable satellite DNA sequences and single nucleotide polymorphisms. Multiple companies offer genotyping services for a small fee.
Cell line stability
Cells grown in culture have a tendency to accumulate genetic and/or phenotypic changes. Studies have shown that long-term culture of hPSCs result in genetic abnormalities including changes at the chromosomal level, which can be detected by karyotyping methods. The karyotypes of human pluripotent stem cell cultures should be frequently tested by a certified cytogenetics laboratory.
Multiple techniques are used to ensure that pluripotent stem cell lines retain their stem cell phenotype, and include expression of stem cell markers, both molecularly and as expressed intracellular or surface markers and the ability to form the three embryonic germ layers. It is highly recommended that several methods be used to monitor cell line stability. Teratoma formation, embryoid body formation, RT-PCR, immunocytochemistry staining and flow cytometry are some of the more common methods employed. While many of these methods are common laboratory techniques it is wise to consider periodic secondary testing through an independent laboratory or core facility when confirmation is required.
As previously stated, the protocols presented here have been thoroughly tested and verified. However, as stem cell research continues to evolve, new practices and protocols are emerging. For example, while feeder-free culture was once a new technology, there are now many standardized feeder-free protocols that can be found. As new techniques arise for more efficient and effective hPSC culture, they certainly should be evaluated and can be adopted after rigorous testing and standardization.
B Reagent preparation
Several reagents must be prepared prior to maintaining human pluripotent stem cells (hPSCs) in culture. It is important to have a firm grasp of the characteristics of each reagent, including components, appropriate preparation, storage and shelf life. This chapter details the preparation of the following reagents necessary for hPSC culture:
Many reagents, especially Knockout Serum Replacer (KOSR), bFGF, and collagenase can have significant variation between lots. When working with a new lot, it is important to compare it directly to the lot that is currently in use, to ensure that the current culture quality and viability are maintained.
B1 Preparation of 0.1% gelatin solution
Reagents
Gelatin, Porcine
Endotoxin-free
Procedure
Prepare 0.1% gelatin solution
Note: Adjust the volume of water and gelatin powder proportionally if a volume other than 500 ml of gelatin is prepared.
Add 0.5 g of gelatin powder to a clean 500 ml pyrex bottle.
Add 500 ml endotoxin-free (e.g., MilliQ) water to the bottle.
Swirl to mix. (At this stage, the gelatin is not soluble).
Autoclave for 30 minutes within 2 hours after mixing.
Cool the 0.1% gelatin solution to room temperature and store at 4–8°C until use. Use this solution within two months of preparation.
B2 Preparation of iMEF culture medium
Supplies
5 ml sterile serological pipets
10 ml sterile serological pipets
25 ml sterile serological pipets
500 ml bottle connected with a 0. 22 μm Stericup
Reagents
DMEM-liquid (Invitrogen 11965-118)
MEM Non-Essential Amino Acid Solution (NEAA) (Invitrogen 11140-050)
Heat Inactivated Fetal Bovine Serum (HI-FBS) (Invitrogen 16000-069)
70% ethanol (Diluted from 95% ethanol, Fisher NC9608803)
Procedure
Preparation of iMEF culture media
In the hood, open a 500 ml filter and bottle unit. Label the bottle:
iMEF and date of preparation
Expiration date (mm/dd/yyy, 14 days after media preparation). For example: Exp: 07/10/2008
Your initials
Add the appropriate amount of ingredients to the 500 ml filter cup as shown in the table below:
Note 1: The DMEM may be measured by pouring directly into the graduated filter cup of the filter/bottle unit. The other solutions should be added using serological pipets.
Note 2: Scale up or down proportionally if another quantity of medium is needed.
Filter the media through the Stericup filter into the attached bottle.
Store the medium bottle at 4°C, and use the medium within 14 days.
B3 Preparation of basic fibroblast growth factor (b-FGF) stock solution
Supplies
1.5 ml or 2 ml sterile microcentrifuge tubes
1000 μl sterile pipette tips
10 ml sterile serological pipets
25 ml sterile serological pipets
50 ml sterile centrifuge tubes
70% ethanol spray
Forceps
Reagents
Basic Fibroblast Growth Factor (b-FGF) (Invitrogen PHG0021)
PBS with 0.01% CaCl2 and 0.01% MgCl2 (Invitrogen 14040-141)
30% BSA (Bovine Serum Albumin) (Sigma A9576)
Procedure
Preparation of 0.1% bovine serum albumin (BSA) solution
Add 0.1 ml of the 30% BSA to 30 ml of PBS.
Cap the tube and mix the solution by inverting 3–4 times. It is now a 0.1% BSA solution.
Preparation of 10 ug/ml b-FGF stock solution
Place one vial of 100 μg lyophilized b-FGF in a 50 ml tube and briefly centrifuge the b-FGF vial at 200 × g for 5 minutes to bring the lyophilized b-FGF to the bottom of the vial.
Use a pair of forceps to carefully remove the vial from the 50 ml tube.
In a biosafety cabinet, carefully remove the cap gently. Avoid contact with any b-FGF powder that has stuck to the cap.
Remove 500 μl of 0.1% BSA solution and add it to the b-FGF vial (Do not touch FGF pellet at the bottom).
Carefully replace the cap.
Carefully remove the cap and gently pipette up and down a few times.
Transfer the b-FGF solution to the 50 ml tube.
Rinse the b-FGF vial with 500 μl solution from the “FGF” tube to collect any residual b-FGF protein in the vial. Return the solution back to the 50 ml tube. Repeat this 2–3 times.
Aliquots of b-FGF (10 ug/ml) stock solution
In the hood, place a set of 50 sterile 1.5 ml or 2 ml vials in a cryo-vial holder.
Aliquot the solution.
Store at −80ºC.
B4 Preparation of pluripotent stem cell culture medium
Supplies
1000 μl sterile pipette tips
20 μl sterile pipette tips
5 ml sterile serological pipets
10 ml sterile serological pipets
25 ml sterile serological pipets
50 ml sterile centrifuge tubes
250 ml Stericup Filter
500 ml Stericup Filter
70% ethanol spray
Reagents
DMEM-F12 media (Invitrogen 11330-032)
Knockout Serum Replacer (KOSR) (Invitrogen 10828-028)
L-glutamine, non-animal, cell culture tested (Sigma G-8540)
MEM Non-Essential Amino acid solution (Invitrogen 11140-050)
Basic Fibroblast Growth Factor (b-FGF); Stock 10 ug/ml
beta-Mercaptoethanol (Sigma M7522)
Procedure
Preparation of culture medium (CM) preparation
Note: Please note that many iPS culture media formulas suggest the inclusion of antibiotics. When working with any cell line, check with the cell line provider for specific media recommendations.
Note: If the final total volume of medium is different from the one listed in the table below, adjust the volume of ingredients proportionally.
In a biosafety cabinet, open a 250 ml or 500 ml filter and bottle unit.
Label the bottle of the filter unit (Culture Medium and date of preparation, expiration date (14 days after media preparation), initials)
According to the final total volume to be prepared, add appropriate amounts of ingredients to the 250 ml or 500 ml filter unit cup as shown in the table below.
Note: The DMEM-F12 may be measured by pouring directly into the graduated filter cup. Other reagents should be added with sterile serological pipets.
Note: When thawing cell lines that have been previously cultured under different conditions, in most cases, it is possible to adapt the cells to the culture medium described here by thawing the cells into this formulation. Thawing hPSCs is fully described in the protocol;
C3 Thawing and Seeding, and replacement of medium for Pluripotent Stem Cells.
Filter the media through the 0.22 μM filter.
Store the medium bottle at 2–8°C, and use the medium within 14 days.
B5 Preparation of collagenase solution
Reagents
DMEM-F12 media
Collagenase Type IV
Procedure
Note: The following procedure is to prepare a collagenase solution at a concentration of 1 mg/ml.
Weigh 100 mg of Collagenase Type IV powder into a weigh boat.
Using a 25 ml pipette, transfer 20 ml of DMEM/F12 medium to the weigh boat containing the collagenase.
Pipette DMEM/F12 medium up and down in the boat to dissolve the collagenase powder. The collagenase should dissolve almost instantly.
When the collagenase is completely dissolved, transfer the solution to a 50 ml tube.
Rinse the residual collagenase in the boat with an additional 20 ml of DMEM/F12 medium and add this to the 50 ml tube containing the collagenase solution, tighten cap.
Spray the 50 ml tube containing the collagenase solution with 70% ethanol, and place the tube in the biosafety cabinet.
To a 150 ml 0.22 μm filter unit pour 60 ml of DMEM/F12 medium and to this add the 40 ml of collagenase solution from the 50 ml tube.
Filter sterilize the collagenase solution (1 mg/ml).
Store the solution at 4°C, and use within 14 days.
References and Suggested Reading
C Cell culture
The protocols provided here describe the standard culture techniques used for pluripotent stem cells. The protocols discussed here have been shown to be reliable and generate reproducible experimental data. The protocols include:
Gelatin Coating of Culture Plates
Thawing and Seeding of Frozen Inactivated Mouse Embryonic Fibroblasts (iMEFs)
Thawing and Seeding of Pluripotent Stem Cells onto a Mouse Embryonic Fibroblast (iMEF) Feeder Layer
Replacement of Medium for Pluripotent Stem Cell Culture
Passaging of Pluripotent Stem Cells on Fresh Mouse Embryonic Fibroblast (iMEF) Plates
Harvesting and Cryopreservation of Pluripotent Stem Cells
C1 Gelatin coating of culture plates
Supplies
6-well tissue culture plates
5 ml sterile serological pipettes
10 ml sterile serological pipettes
Reagents
0.1% gelatin solution
Procedure
Coat culture plates using 0.1% gelatin solution
Warm to room temperature an appropriate amount of gelatin solution. For 6-well plates, use 2 ml of 0.1% gelatin solution for each well.
Place the plates that are to be coated in the biosafety cabinet.
Label the cover of the plate (not over the wells) with: “G” for gelatin, date, initials
Add 2 ml of 0.1% gelatin solution to each well.
Tilt or swirl the plates in several directions so that the liquid covers the entire surface area.
Place the plates in a 37°C incubator.
Note: The plates will be ready for use in 4 hours. They can be used for up to 7 days.
C2 Thawing and seeding of frozen inactivated mouse embryonic fibroblasts (iMEFs)
Supplies
5 ml sterile serological pipettes
10 ml sterile serological pipettes
25 ml sterile serological pipettes
15 ml sterile centrifuge tubes
50 ml sterile centrifuge tubes
70% ethanol spray
Reagents
iMEF culture medium
Gelatin-coated plates
Frozen iMEFs
Procedure
Prepare gelatin plate(s) in the biosafety cabinet
Remove gelatin plate(s) from the incubator and place inside the biosafety cabinet.
Aspirate the gelatin solution from the plate(s) completely with a Pasteur pipette.
Return the plate(s) to the incubator for later use.
Thaw iMEFs
Note 1: To seed 6 wells in a 6-well plate, 1.2–1.5 × 106 iMEFs are required (assuming a 90% recovery of iMEFs from one freezing and thawing cycle).
Note 2: Thaw no more than two vials at a time.
Note 3: Wear eye protective safety glasses and insulated gloves when removing cryovials from the liquid nitrogen freezer.
Bring an appropriate amount of MEF medium (iMEF CM) to 37°C (assuming 2 ml per well + 9 ml for thawing)
Place the warmed iMEF CM in the biosafety cabinet after thoroughly cleaning the outside of the container with 70% ethanol.
Label a sterile tube (both on the cap and side) in the biosafety cabinet as “iMEF”.
Transfer 9 ml iMEF CM into the “iMEF” tube.
Open the LN2 freezer and take out the correct vial(s) of iMEFs.
Warm the vial(s) slightly between gloved hands while walking to the water bath.
In the 37°C water bath, immerse the vial in the water without submerging the cap. Swirl the vial gently. After about 30 seconds, check frequently to see if the frozen solution is beginning to melt. This process may take up to 1 minute.
When there is only a small piece of ice floating in the vial, remove the cryovials from the water bath.
Place the vial(s) in the biosafety cabinet after thoroughly cleaning the outside of the cryovials with 70% ethanol.
In the biosafety cabinet, pipette the cells up and down gently a couple times, and then transfer the cell suspension drop-wise to the tube labeled “iMEF” while gently swirling the tube. This will help to reduce osmotic shock to the cells.
Centrifuge for 5 minutes at 200 × g.
Return the 50 ml centrifuge tube to the biosafety cabinet after spraying it with 70% ethanol.
Aspirate the supernatant from the tube. Be careful not to touch the cell pellet.
Gently flick the bottom of the tube to loosen the cell pellet.
Add 10 ml of iMEF CM to resuspend and mix the cells by pipetting gently up and down a few times.
If necessary, count iMEFs.
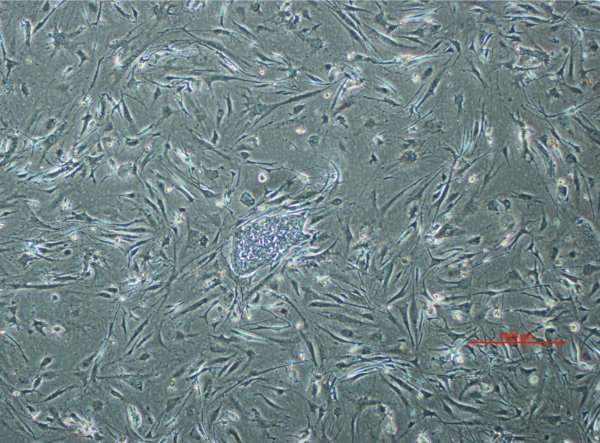
Add iMEF CM to the iMEF tube for a final cell concentration of 0.1–0.13 × 106 iMEFs/ml. Image shows correct concentration of MEFs.
Seed iMEFs in 6-well plate
Take the gelatin plate(s) from the incubator to the biosafety cabinet. Label the plates with “iMEF”, date and initials.
Resuspend the iMEFs completely by gently pipetting up and down a few times and add 2 ml/well to gelatin coated wells.
When all plates in the biosafety cabinet are seeded, slide the plates back and forth and side to-side (cross motion) three to five times inside the biosafety cabinet to evenly distribute cells and make another 3–5 cross-motions with the plates when transferred to the incubator.
Post seeding
Do not disturb the freshly seeded plates for at least the next 12 hours.
Check each plate of cells under the microscope the following day for MEF quality and for signs of contamination
Record observations
Repeat the above each day until the plates are used for pluripotent stem cell culture.
Note: Discard unused or contaminated iMEF plates after 5 days
C3 Thawing and seeding, and replacement of medium for pluripotent stem cells
Supplies
5 ml sterile serological pipettes
10 ml sterile serological pipettes
25 ml sterile serological pipettes
15 ml sterile centrifuge tubes
50 ml sterile centrifuge tubes
70% ethanol spray
Reagents
Monitor iMEFs under microscope
Observe the number of plates needed for this protocol one under a microscope.
Return the plates to the incubator if there is no contamination.
Note: If there is contamination in any culture plate, disinfect all wells in the plate(s) with bleach (whether contaminated or not).
Thaw pluripotent stem cells
Note 1: Wear eye protective safety glasses and insulated gloves when removing cryovials from the liquid nitrogen freezer.
Note 2: Never thaw cells simultaneously from different stem cell lines. If they are from the same line and same lot, thaw no more than two vials at a time in order to minimize prolonged exposure of thawed cells to cryopreservation medium.
Bring an appropriate amount of DMEM-F12 and CM to room temperature. Place them in the biosafety cabinet after cleaning with 70% ethanol.
Place the warmed DMEM-F12 and CM aliquots in the biosafety cabinet after thoroughly cleaning with 70% ethanol.
Label a sterile tube (both on the cap and side) in the biosafety cabinet as “cells”.
Transfer 9 ml CM per cryovial of hPSCs into the “cells” tube.
Remove the cell vial(s) from the LN2 freezer and verify the label on the cryovial(s).
Warm the vial(s) slightly between gloved hands while walking to the water bath.
In the 37°C water bath, immerse the vial(s) in the water without submerging the cap. Swirl the vial gently.
After 30 seconds, check frequently to see whether frozen medium is beginning to melt. This process may take up to 1 minute.
When there is only a small piece of ice floating in the cryovial(s), remove the vials(s) from the water bath.
Place the vial(s) in the cryovial holder (if using) after thoroughly cleaning with 70% ethanol.
Pipette the cells gently up and down a few times and transfer the cell suspension drop-wise to the CM in the labeled “cell” tube while gently swirling the medium to reduce osmotic shock to the cells.
Centrifuge for 5 minutes at 200 × g.
During the centrifugation period, take the approved iMEF plates from the incubator and place them in the biosafety cabinet:
Label the plate(s) in the biosafety cabinet (the date, the words “thaw” and the cell line ID, the passage number plus one, and initials)
When centrifugation is complete, return the centrifuge tube to the biosafety cabinet after cleaning with 70% ethanol.
Aspirate the supernatant from the tube (be careful not to touch the cell pellet).
Note: Normally, one vial of thawed hPSCs will be seeded in one well of a 6-well plate.
Add the 2.5 ml of CM to the tube and pipette cells gently a several times to mix.
Seed pluripotent stem cells in 6-well plates
Note: To avoid mislabeling cells and cross-contamination, seed cells from different lines in different iMEF plates.
Aspirate the iMEF medium completely from the iMEF plates with a Pasteur pipette.
Rinse and aspirate the wells with 2 ml/well of DMEM-F12.
Mix the cells in the tube completely by pipetting gently up and down a few times, and drop-wise add cells at 1 vial per well.
When all plates are seeded, slide the plates in back and forth and side-to-side (cross-motion) three to five times inside the biosafety cabinet. Make another 3–5 cross-motions when transferred to the incubator.
Post seeding
Do not move the freshly seeded plates for at least the next 12 hours.
Note: From here on, monitor cell growth and starting on day 2, change medium daily. Colonies may not be visible for a few days.
Note: When thawing cell lines that have been previously cultured under different conditions, in most cases, it is possible to adapt the cells to the culture medium described in these protocol by directly thawing the cells into it. The complete formulation for culture medium is provided in the protocl;
B4 Preparation of Pluripotent Stem Cell Culture Medium.
C4 Replacement of pluripotent stem cell culture medium
Procedure
Monitor hPSC growth
Take the hPSC plates from the incubator one by one and evaluate cell growth under the microscope.
Return the culture plates to the incubator.
Note: If there is contamination in any culture plate, do the following:
If no further culturing will be done with the plate, discard all contaminated culture wells/plates with at least 250 μl bleach (per well) for at least 30 minutes.
If there are uncontaminated wells in a plate with contamination are to be cultured further, then perform the following:
Move the plate to a biosafety cabinet and disinfect each contaminated well with 250 μl bleach for 5 minutes.
After removing the disinfected content from the well completely, change medium for other wells and place the plate in a specifically designated incubator for observation during the next few days.
Once the culture is free of contamination for five days, the plate can be returned to regular incubator for further culture.
Change medium for hPSC culture
Warm an appropriate amount of CM to room temperature (For a 6 well plate, 2.5 ml of CM is needed per well).
Place the warm “CM” tube or bottle in the biosafety cabinet after thoroughly cleaning with 70% ethanol.
Take the hPSC plates from the incubator and place them in the biosafety cabinet.
Aspirate the spent medium completely using a Pasteur pipette.
Add 2.5 ml fresh CM to each well in a 6-well plate.
Return the hPSC plates to the incubator.
Note: Repeat the above procedure daily until the cells are ready for passage or splitting to new iMEF plates.
C5 Passaging of pluripotent stem cells on fresh iMEF plates
Supplies
5 ml sterile serological pipettes
5 ml sterile glass pipettes
10 ml sterile serological pipettes
15 ml centrifuge tubes
50 ml centrifuge tubes
Colony marker
Reagents
DMEM-F12
Collagenase Solution
hPSC Culture Medium
6-well plates with iMEFs
Preparation
Determine when and how to passage (or split) pluripotent stem cells
Note: Different cell lines have different growth kinetics. Therefore, different splitting schedules may be used for different cell lines. However, it is generally true that hPSCs grow slowly during the first couple of weeks after being thawed, then faster until the growth rate reaches a plateau. The cell growth rate will then stay in that plateau for many passages if cells are cultured properly. It is essential to observe cell growth daily to become familiar with the growth kinetics of the cell line being cultured.
Under a microscope, carefully evaluate cell growth and quality. Observations will be used to determine when and how often the cells are passaged.
Healthy, undifferentiated hPSC colonies generally have well-defined uniform borders and the individual cells within the colony appear to be similar. The exact colony morphology will differ with different cell lines and culture conditions (i.e., whether iMEFs, mTeSR1, or conditioned medium with Matrigel are used for hPSC culture).
Spontaneously differentiated cells may resemble cobblestones or trophoblast-like cells. The differentiated cells may reside at the colony perimeter or in the center of the colony. There can also be differentiated colonies containing large clumps of swirled tissue, or little balls of cells, like embryoid bodies. Different culture conditions yield different types of differentiated cells.
In general, passage cells when any of the following occur:
iMEF feeder layer is two weeks old
50% of the colonies are (greater than 2000 μm)
Colonies are too dense (at approximately 70% confluence)
Colonies exhibit increased differentiation.
Determine how to passage (or split) pluripotent stem cells
The method of choice depends on the differentiation level in the cultures and how many colonies to be passaged.
If differentiation level is low (less than 20%), the method of PTD-Enzyme is recommended.
If differentiation level is high (more than 70%), the method of PTK is recommended.
If differentiation level is
intermediate (20–70%), either method can be chosen depending on how many good colonies are available and how many colonies are to be passaged:
If there are many undifferentiated colonies available and only limited colonies need to be passaged, PTK is recommended.
If there are many undifferentiated colonies available and as many good colonies as possible need to be passaged, PTD-Enzyme is recommended.
If there are a limited number of undifferentiated colonies available and as many good colonies as possible need to be passaged, either method is appropriate
Determine the splitting ratio to be used.
Look at records from the previous passages (up to three passages). Compare the cell splitting-ratios and the colony confluence levels at the harvest days between sequential passages.
If cells grew significantly faster than the previous passage, increase the splitting ratio.
If the growth rate is similar to the previous passage, use the same cell splitting-ratio.
If cells grew significantly slower than the previous passage, split cells at a lower ratio than the previous one.
Note: Regardless of the method used, it is prudent to set aside one well of cells from the same culture used for passaging for an additional 1–2 days in case of errors or contamination during passaging.
Calculate how much medium and solution are needed for this process
For harvesting and plating hPSCs on iMEFs, the following are required:
DMEM-F12 (2 ml per well for each iMEF Plate)
Collagenase solution (1 ml per well to be harvested)
Culture medium (2.5 ml per well for harvest; 2.5 ml per well for seeding + extra)
Calculate and record the amount of the above materials required for this process
Evaluate & prepare fresh iMEF plates
Take the freshly seeded iMEF cell plates from the incubator one at a time and evaluate cell culture under microscope.
Return the plates to the incubator if there is no visible contamination.
Take the approved iMEF plates and place them in the biosafety cabinet.
Label the plates with the cell line name, new passage number, the date and initials.
Completely aspirate the medium from the iMEF plate(s) with a Pasteur pipette.
Add 2 ml/well of fresh DMEM-F12 medium.
Gently swirl the plate to rinse the well walls.
Aspirate the DMEM-F12 medium completely from the plate(s) with a Pasteur pipette.
Add 1.5 ml of fresh culture medium/well.
Return the plate(s) to the incubator.
Procedure-pick to discard-enzyme Harvest cells (no more than two plates at a time)
Label a sterile tube as “Cells” for collecting hPSCs later.
Take the hPSC plate(s) from the incubator and evaluate under a microscope. If using a colony marker, mark the differentiated colonies on the bottom of the plate and place in the biosafety cabinet.
Using a Pasteur pipette, aspirate all marked (differentiated) cells in the plate by touching the colonies with the tip of the pipette. The differentiated areas will be sucked away leaving the undifferentiated colonies in the plate.
Aspirate the remaining medium along with the floating differentiated cells or cell debris using a straight Pasteur pipette.
Set the timer at 3 minutes and 10 minutes, respectively.
Add 1 ml collagenase/well and return the plate(s) to the incubator. Start the timer
After 3 minutes of incubation, check the plate(s) under a microscope to determine whether the cell colonies are ready to be harvested. If they are ready, the perimeter of the colony should appear folded back, separating from the iMEFs. If they are not ready, return the plate back to the 37°C incubator for another 3–5 minutes of incubation.
Note: Incubation length depends on the freshness of the collagenase solution. The older it is, the longer the incubation time will be. However, do not exceed 15 minutes of incubation time.
When collagenase incubation is complete, aspirate collagenase from the wells in the biosafety cabinet. Be careful not to disturb the cell/colony layer.
Add 2 ml culture medium to each well in the plate(s).
Using a sterile 5 ml glass pipette, take up most of the medium from one well. Use the pipette to scrape the cells/colonies in the well while slowly releasing the medium into the well to wash the cells/colonies off the surface. Pipette up and down gently to minimize bubbles. Repeat these steps until most of the cells/colonies are detached from the surface. Leave the contents in the well until cells in all of the wells are detached.
When cells in all wells of one plate are detached, transfer the cell/colony solution from all wells into the labeled sterile tube. Rinse all the wells in the plate with another 3 mls of CM. Transfer the cell/colony solution to the same tube. If there are more plates, harvest remaining cells into the same tube.
Spin and disrupt colonies
Centrifuge cells at 200 × g for 5 minutes.
Aspirate the supernatant from the tube (do not touch the cell pellet).
Resuspend the cell pellet in an appropriate volume of culture medium (1 ml/well to be seeded, plus 0.5 ml extra for pipetting error).
Using a 5 ml glass pipette, disrupt the cell clusters by pipetting cells up and down gently a few times. Pipette carefully to avoid bubbles by keeping the pipette tip in the solution at all times.
Note: In the above step, pipette the cell suspension up and down relatively fast, initially, to disturb big cell clusters. Check frequently to see if the big cell clusters are disrupted. When most of the colonies are small enough to remain suspended in solution, the cells are ready to be plated. When mixing, pipette slowly as not to break down clusters any more than needed.
Add cells to iMEF plates (processing no more than three plates at a time):
Return the iMEF plates (containing 1.5 ml/well culture medium) to the biosafety cabinet.
Using a 5 ml glass pipette, gently pipette cells up and down a few times. Then take 3 ml of the cell suspension and drop-wise (in a circular motion) add 1 ml/well to each of the three wells.
Repeat the above two steps until cells are distributed to all wells in the iMEF plate.
When complete, slide the plates in a cross-motion along the inside surface of the biosafety cabinet. Make another 3–5 cross-motions when transferred to the incubator.
Note: Do not move the freshly seeded plates for at least the next 12 hours. Check cells under microscope the next day.
Procedure-pick to keep Prepare sterile bent pasteur glass
Note: Bent Pasteur pipettes are required to remove undifferentiated hPSCs for transfer to new iMEF plates.
Take a 9-inch Pasteur glass pipette and heat the area about two inches from the tip briefly over the open flame of an alcohol burner (about 3–5 seconds).
Once the heated area is pliable, remove the pipette from the flame and gently pull the tip at a 45º angle until it separates from the base of the pipette. Place the tip briefly over the open flame to form a small ball. Ideally, the bent pipette should now have a small 0.5-inch hook that ends with a small ball at the tip.
Repeat steps above to make additional bent Pasteur pipettes and place them in a Pasteur pipette container.
Autoclave bent Pasteur glass pipettes prior to use.
Transfer areas of undifferentiated cells to new iMEF plates (Pick to keep)
Note: This section describes PTK at a ratio of 1:1 (1 well to 1 well). If there are few viable colonies, a ratio of 2:1 or higher may be needed.
Remove a hPSC plate from incubator and place it in a biosafety cabinet.
Aspirate the spent medium from the wells with a sterile straight Pasteur pipette.
Add 1.5 ml fresh culture medium to each well containing cells.
Mark the undifferentiated area in the wells on the plate bottom using a colony marker attached to an inverted microscope.
Transfer the plate to the biosafety cabinet. Detach the undifferentiated cells in the well with a modified/bent pipette. Pipette the cell clusters a few times to break them into aggregates.
Transfer the freshly prepared iMEF plate containing 1 ml/well culture medium from the incubator and place it in the biosafety cabinet.
From the stem cell plate, gently transfer the medium (about 1.5 ml) along with the floating colony pieces into the freshly prepared iMEF well at 1:1 ratio (one well to one well).
Return the seeded plate to the incubator. Slide the plate in a cross-motion on the shelf of the incubator 3–5 times.
Add 2.5 ml of medium to the well of the old plate and return it to the incubator for 1–2 days of additional culture.
Note 1: If the need arises to open the incubator while the cells are attaching to the surface (during the next 12 hours), open and close the incubator door gently.
Note 2: Replace culture medium daily.
C6 Harvesting and cryopreservation of pluripotent stem cells
Supplies
5 ml sterile glass pipettes
5 ml sterile serological pipettes
10 ml sterile serological pipettes
15 ml sterile centrifuge tubes
50 ml sterile centrifuge tubes
Cryovials (Nalgene 5000-0020)
Mr. Frosty™ (Nalgene® 5100-0001)
Procedure
Determine when to harvest cells
Note: Harvest cells when they are in the log phase of growth, which is normally between 4 to 6 days after plating.
Under a microscope, evaluate cell growth and quality carefully.
All of the following observations will be used to determine when cells are ready to be harvested. Harvest cells when:
There is less than 10% differentiation in the previous passage
Colonies of cells reach 70% confluence.
iMEF feeder cells in the culture are less than two weeks old.
About 50% of colonies are greater than 2000 μm
Prepare cryovials
Make sure there are sufficient freezing containers on the bench near the biosafety cabinet for freezing cells. Make sure all freezing containers have a sufficient amount of fresh (made on the day of harvest) isopropanol in them. Each freezing container can hold 18 cryovials.
Label each cryovial (Provider's cell line name and passage number, date, initials) and expose to UV light for at least 30 minutes.
Calculate how much culture medium and collagenase solution is needed for this process
For harvesting hPSC, the following are required:
Note: Do not place collagenase in water bath to warm.
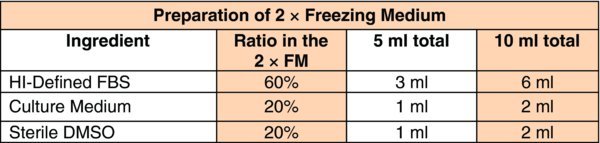
Prepare 2× cryopreservation media (or freezing medium, FM) in the biosafety cabinet
Note: Prepare 2 × FM media just before use (fresh).
Determine the amount of cryopreservation media required:
According to the volume calculated above, label one sterile 50 ml conical tube or a larger size sterile bottle as 2 × FM (on both the cap and side).
Add the following, proportionately, to the labeled container:
Mix and filter through a 0.22 μM filter unit
Keep the above solution on ice or in a 4–8°C refrigerator and use it within the day.
Note: This 2× FM will be added to cell suspension at a 1:1 volume ratio, resulting in 1× freezing medium with a final concentration of 10% DMSO and 30% FBS, respectively.
Incubate cells with collagenase solution (prepare no more than two plates at a time):
Place the warm culture medium aliquot in the biosafety cabinet after thoroughly cleaning with 70% alcohol.
Label a sterile container as “Cells” for collecting hPSCs.
Set the timer at 3 minutes and 5 minutes, respectively.
Take out one or two plate(s) from the incubator and place it/them in the biosafety cabinet.
Aspirate the medium completely from the plate(s) using a Pasteur pipette.
Add 1 ml collagenase/well.
Return the plate(s) to the incubator.
Start the timer for 3 minutes
After 3 minutes of incubation, check the plate(s) under a microscope to determine whether the cell colonies are ready to be harvested. When they are ready, the perimeter of the colony should appear folded back, separating from the iMEFs. If they are not ready, return the plate back to the 37ºC incubator for another 2–3 minutes of incubation.
Note: Incubation time depends on the freshness of the collagenase solution. The older it is, the longer the incubation will take. However, do not exceed 15 minutes.
When collagenase incubation is complete, place the plate in the biosafety cabinet.
Aspirate collagenase from the wells. Be careful not to disturb the cell/colony layer.
Harvest cells
Note 1: HPSCs recover better from freezing and thawing when frozen in large aggregates.
Note 2: Leave the contents in the well until all wells are scraped.
Add 2 ml culture medium to each well.
Using a 5 ml glass pipette, take up most of the medium from one well. Scrape the cells/colonies in the well using the pipette while slowly releasing the medium into the well to wash the cells/colonies off the surface. Gently pipette the medium up and down during the wash to minimize bubbles and to disrupt cell clumps. Repeat these steps until most of the cells/colonies are detached from the surface.
Detach cells/colonies in the other wells by repeating the above steps.
When all wells are complete, combine the cell/colony solution from all wells into the bottom of the labeled sterile “Cells” container. Rinse all of the wells in one plate with another 3 ml of culture medium. Transfer the cell/colony solution to the same sterile “Cells” container.
Freeze cells
Note 1: Once in DMSO freezing medium, cells should be frozen as quickly as possible (within 5 minutes).
Note 2: When freezing a large lot, different parts of the freezing process may be completed by different personnel in order to reduce the time that cells are in the freezing medium.
Centrifuge cells at 200 × g for 5 minutes.
During the centrifugation period, loosen all cryovial caps in the biosafety cabinet without taking them off.
When centrifugation is complete, return “Cells” container to the biosafety cabinet after cleaning with 70% ethanol.
Aspirate the supernatant off the cell pellet.
Resuspend the cell pellet gently in appropriate volume of culture medium (0.5 ml/well plus 0.5 ml extra).
After cleaning with 70% ethanol, place the cold 2 × freezing medium (FM) in the biosafety cabinet.
Dropwise, add an equal volume of 2 × FM to the cell suspension.
After pipetting to gently to mix the cells, transfer cells to cryovials at 1 ml/vial.
Transfer cryovials to Mr. Frosty freezing container and place the freezing container in a –80°C freezer overnight.
Note: Cells need to be cryopreserved in LN2 freezers.
The next day, quickly transfer the cryovials into the LN2 freezer on dry ice.
D References
- 1.
Yao S, Chen S, Clark J, Hao E, Beattie GM, Hayek A, Ding S. Long-term self-renewal and directed differentiation of human embryonic stem cells in chemically defined conditions.
Proc Natl Acad Sci U S A. 2006;103:6907–12. [
PMC free article: PMC1458992] [
PubMed: 16632596] [
CrossRef]
- 2.
- 3.
Chase LG, Firpo MT. Development of serum-free culture systems for human embryonic stem cells.
Curr Opin Chem Biol. 2007;11:367–372. [
PubMed: 17692558] [
CrossRef]
- 4.
Upoff C, Drexler H. Elimination of Mycoplasmas from Infected Cell Lines Using Antibiotics in Cancer Cell Culture Methods in Molecular Biology™
2011;731:105–114. [
PubMed: 21516401] [
CrossRef]
- 5.
Nicklas W, Kraft V, Meyer B. Contamination of transplantable tumors, cell lines, and monoclonal antibodies with rodent viruses.
Lab Anim Sci. 1993;43:296–300. [
PubMed: 8231085]
- 6.
Stacey GN, Cobo F, Nieto A, et al. The development of ‘feeder’ cells for the preparation of clinical grade hES cell lines: challenges and solutions.
J Biotechnol. 2006;125:583–588. [
PubMed: 16690155] [
CrossRef]
- 7.
Nelson-Rees WA, Daniels DW, Flandermeyer RR. Cross-contamination of cells in culture.
Science. 1981;212:446–452. [
PubMed: 6451928] [
CrossRef]
- 8.
MacLeod RA, Dirks WG, Matsuo Y, et al. Widespread intraspecies cross-contamination of human tumor cell lines arising at source.
Int J Cancer. 1999;83:555–563. [
PubMed: 10508494]
General
Freshney, Ian R. Culture of Animal Cells: A Manual of Basic Technique, 5th edition. John Wiley & Sons, Inc. Hoboken, New Jersey, 2005: 192–195.
Passaging
Gonzalez R., Wesselschmidt R.L. Schwartz P.H. & Loring J. Human Embryonic Stem Cell Culture in Human Stem Cell Manual: A Laboratory Guide, ed Loring J., Wesselschmidt R.L., & Schwartz P.H.NY: 2007;5–7.
Harvest and cryo
Katkov II, Kim MS, Bajpai R, Altman YS, Mercola M, Loring JF, Terskikh AV, Snyder EY, Levine F (2006). Cryopreservation by slow cooling with DMSO diminished production of Oct-4 pluripotency marker in human embryonic stem cells. Cryobiology 53: 194–205.
Ware CB, Nelson AM, Blau CA (2005). Controlled-rate freezing of human ES cells. Biotechniques 38: 879–880, 882–873.
- *
Last revised March 28, 2012. Published June 10, 2012. This chapter should be cited as: Borowski M., Giovino-Doherty M., Ji L., Shi M.-J., Smith K.P., and Laning J. Basic pluripotent stem cell culture protocols (June 10, 2012), StemBook, ed. The Stem Cell Research Community, StemBook, doi/10.3824/stembook.1.63.1, http://www.stembook.org.