

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006.

The coding region of the α-galactosidase A gene (GLA) consists of 1290 base pairs, is divided into seven exons and defines a polypeptide of 429 amino acids. The great majority of disease-related GLA mutations are unique ('private'). We have compiled a list of 429 mutations of the GLA gene from the published literature, including 306 point mutations (missense, nonsense and those affecting splice sites), 115 'short-length' rearrangements (affecting fewer than 60 nucleotides) and eight gross rearrangements (affecting one or more exons). Based on the number of different changes at any nucleotide position, there is no obvious 'hot spot' for point mutations, although mutations of CpG dinucleotides account for the majority of recurrent point mutations seen in unrelated families with Fabry disease. Remarkably, about one-third of the short-length rearrangements occur in exon 7, which accounts for only 22% of the coding region, suggesting that this part of the gene is susceptible to rearrangement. The recent elaboration of a putative three-dimensional structure of α-galactosidase A by X-ray crystallography may provide a better insight into how the enzyme works at the molecular level. This knowledge has recently been used for computer modelling of the structure of α-galactosidase A mutants and may result in improved understanding of the molecular pathology of the mutated protein. It is hoped that an increased understanding of structure/function correlates will help to develop alternative therapies or adjuvant treatments for Fabry disease.
Introduction
This chapter gives a short overview of the α-galactosidase A gene (GLA), which is mutated in Fabry disease, and the mutations found in the gene in health and disease. The term mutation refers to a permanent alteration of the genetic material. In daily practice, however, the word mutation is frequently (but incorrectly) associated only with a disease-causing effect. Due to this negative connotation, the neutral terms sequence variant, sequence alteration or allelic variant are used frequently in current literature.
Classification and nomenclature of mutations
From the point of view of pathology, only mutations that affect the phenotype (in a broad sense) are of relevance. These mutations can be divided into those that cause disease and those that are non-pathogenic. Clearly, non-pathogenic mutations may either be expressed phenotypically or be silent. From the point of view of genetics, mutations can be: familial – that is, transmitted from generation to generation within a family; or de novo, with a change first being detected in the index case. In terms of the change in the genetic material, we usually distinguish between novel mutations that have not been previously reported, and those that have already been documented in the literature. The fact that the same mutation has been found in two different patients may indicate either that the two individuals are (distantly) related or that we are dealing with recurrent mutations due to de novo events – that is, the same mutation has occurred independently on two occasions.
The prospect of computer-based mutation databases requires a uniform and unequivocal assignment of sequence variants. Following the initiative of a small group of human geneticists, recommendations have been put forward during the past decade to establish a standard nomenclature for describing mutations. Today, the majority of those working in the field largely agree on the 'Nomenclature for the description of sequence variations' that is posted on the HGV (Human Genome Variation) Society web page (http://www.genomic.unimelb.edu.au/mdi/). In view of the fact that mutation nomenclature is a specialized domain of human genetics that most readers of this book may not be familiar with, we have compiled a brief description of the currently used nomenclature.
In general, the abbreviated names (acronyms) of human genes are written in italic capital letters. This way, one can distinguish between the gene and its product, which is written in a regular font. For DNA, the capital letters A, C, G and T are used, corresponding to the nucleotides adenine, cytosine, guanine and thymine, respectively, whereas the three-letter amino acid code is preferred when describing a change at the protein level. The convention is that designation of nucleotide (DNA) changes begins with a number, and designation of protein changes with a letter. Thus, 100C>T (cytosine is replaced by thymine at position 100) is a nucleotide change, whereas C100T (more correctly Cys100Thr) is a missense mutation (cysteine is replaced by threonine). The recommended designation helps to avoid confusion about whether A, C, G or T represent nucleotides or amino acids, which may occur if a shorthand (one-letter amino acid code) abbreviation is used. The description of any sequence change is always preceded by a letter indicating the type of sequence referred to:
- 'g' for genomic DNA
- 'c' for cDNA (complementary DNA)
- 'p' for protein.
Positions of amino acids and nucleotides (cDNA) are numbered starting, respectively, with the initiation methionine and the nucleotide A of the ATG-translation initiation triplet as number 1. For numbering nucleotides in introns (the intervening sequences between exons), the last nucleotide of the preceding exon (exons are the coding portions of the gene) and a plus sign are used at the beginning of the intron, such as 639+1G>A. Likewise, the designation consisting of the first nucleotide of the following exon and a minus sign correspond to the end of the intron, for example 640–1G>T. In this chapter, DNA mutations are described according to the GLA cDNA sequence GenBank U78027.1 or to the genomic sequence GenBank X14448.1 (http://www.ncbi.nlm.nih.gov/Genbank/).
Polymorphisms and rare sequence variants of the GLA gene
The term polymorphism refers to the existence of more than one normal allele at a gene locus, with a frequency of the minor (rare) allele greater than 1% in the normal population. If the frequency of the minor allele is less than 1%, it is referred to as a rare variant. Table 1 shows a list of common GLA gene polymorphisms and the frequencies of the minor (rare) alleles. The differences seen between unaffected controls and patients/carriers for Fabry disease, as determined in our laboratory, are probably due to the small sample sizes. Remarkably, the only coding variant (c.937G>T, p.Asp313Tyr) was detected about ten times more frequently (approximately 5%) in our cohort than the frequency of 0.45% reported originally [1].
The GLA gene and its mutations in Fabry disease
The GLA gene was mapped to the region q22.1 of the X chromosome. The coding part of the gene consists of 1290 base pairs (bp), is divided into seven exons, ranging in size from 92 to 291 bp, and defines a polypeptide of 429 amino acids. As expected for an X-chromosomal trait with reduced reproductive fitness of patients and increased frequency of spontaneous (de novo) mutations, most of the patients/families have different mutations; that is, the great majority of GLA mutations are unique ('private'). There are only a few reports of male patients carrying two different, and most likely disease-causing, GLA mutations on the same allele, such as p.Glu66Gln and p.Arg112Cys, or p.Leu89Arg and a 1 bp deletion in codon 303 [2, 3]. In contrast, about 5% of the patients carry a pathogenic GLA mutation and the non-disease-associated variant p.Asp313Tyr.
We have compiled a list of 429 GLA mutations published in the literature by ourselves and others (as of 31 December 2005). Figures 1 and 2 show the distribution of mutations over the seven coding exons, grouped according to the nature of the mutation; that is, point mutations (missense, n = 240, 55.9%; nonsense, n = 48, 11.2%; and those affecting splice sites, n = 18, 4.2%) and 'short-length' rearrangements (affecting fewer than 60 nucleotides, n = 115, 26.8%), whereas Table 2 lists the large rearrangements (n = 8; 1.9%) described to date.

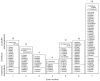
Table 2
Larger rearrangements of the α-galactosidase A gene affecting one or several exons. Mutations that are indented in the left-hand column have been characterized at the nucleotide level.
The relative frequencies of different classes of mutations identified in patients registered in FOS – the Fabry Outcome Survey [4] –agree very well with the above data collected from the literature.
Based on the number of different changes at any nucleotide position, there is no obvious mutation 'hot spot', when considering all 288 point mutations (missense and nonsense) scattered over the 1290 bp coding region. However, there are some differences in the gross distribution of the different mutations. The data show a clustering of point mutations in exon 5, in which a total of 51 point mutations have been reported to date. Given the size of exon 5 (162 bp, comprising 12.6% of the coding sequence) and the observation that it harbours 17.7% of all point mutations in the coding region shown in Figure 1, one can conclude that, of the seven GLA exons, exon 5 has the highest relative frequency of point mutations (3.15/10 bp), followed by exon 6 (198 bp and 56 point mutations; 2.83/10 bp) and exon 3 (178 bp and 45 point mutations; 2.53/10 bp). In total, exons 3, 5 and 6, which comprise 41.7% of the coding sequence, harbour 152 of the 288 point mutations (52.8%). CpG dinucleotides are known to be prone to point mutations due to methylation-induced deamination of 5-methyl cytosine. Of the 14 CpGs in the GLA coding sequence, point mutations have been described in ten (no point mutations of the CpG dinucleotides have yet been found in codons 39, 118, 315/316 and 367/368). CpG-associated mutations account for the majority of recurrent point mutations seen in families with Fabry disease (Table 3).
Table 3
Recurrent point mutations in the α-galactosidase A gene published in the literature.
About one-third (39 out of 115) of the small rearrangements occur in exon 7, which accounts for about 22% of the coding region. This clustering of mutations suggests that this part of the gene is susceptible to rearrangements. It has recently been recommended that the term 'dup' (duplication) is used when the mutation creates a run of two or more bases. Compared with the earlier designation 'ins' or 'insertion' for this type of sequence extension, 'dup' seems to be simpler, and prevents confusion regarding the exact position introduced. For rearrangements in general, the most 3′ (toward the end of the gene) position possible should be arbitrarily assigned to have been changed. In Figure 2, we have followed these most recent recommendations of the Nomenclature Committee of the HGV Society.
Analysis of different mutation entries in databases and in the literature shows that a standard nomenclature is essential to compare data obtained in various laboratories; for example, to determine whether a given mutation has already been reported by others. Figure 3 presents a typical example. A 3 bp deletion (GAG) at position c.1072 has been reported by Blanch et al. [5], whereas Shabbeer et al. [6] reported a 3 bp deletion (AGG) at position c.1070. As shown in Figure 3, the two mutations seem to be (structurally) identical.
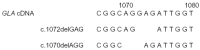
If two mutations can be described by the same formula, we might assume that we are dealing with the same molecular change in both cases. However, this is not necessarily true, as it has been well documented that the same mutation might occur several times independently (recurrent mutation). Disregarding this latter possibility may lead to an underestimation of the natural variability of the mutation spectrum and obscure the fact that certain parts of the GLA gene are prone to mutations. An independent origin of recurrent mutations can be assumed if family analysis suggests that the mutation occurred de novo. In the cohort of patients in FOS, we found two male cases with the p.Gln157X mutation, both from Germany, in which the patients' mothers did not carry the mutation; that is, it appeared to arise de novo. Furthermore, a recurrent event can be assumed if two patients with Fabry disease carry the same GLA mutation on a different genetic background. To examine this question, an individual pattern of polymorphisms, which consists of a number of different genetic variants and is referred to as the haplotype, is defined for each of the patients under study. Table 1 shows the haplotypes of two German male patients, both carrying the c.1019G>A (p.Trp340X) mutation, for a total of five GLA DNA polymorphisms. In view of the fact that the two patients carry different alleles for the intron 4 and intron 6 SNPs (single nucleotide polymorphisms), it is likely that the two probands are unrelated and that this particular mutation has occurred independently on two occasions.
The frequency of de novo mutations in Fabry disease is unknown. Based on theoretical considerations, we have speculated that it might be approximately 3–10% of all cases [7]. Nine cases of molecularly proven de novo (novel) point mutations have been reported to date by ourselves and other investigators (Table 4) and an individual carrying a (recurrent) de novo p.Gln157X mutation has recently been detected in our laboratory (E Schäfer and A Gal, unpublished observation). The total number of different GLA point mutations compiled in this review is 306. Conservatively, we can assume that all but the ten de novo mutations were familial. In this case, the available data suggest that the minimum proportion of new mutations in the cohort studied here is 3.3% (10/306), which is similar to the figure (mentioned above) that has been derived from theoretical considerations.
Table 4
De novo point mutations of the α-galactosidase A gene published in the literature.
The gene product
Recently, Garman and Garboczi [8] published the structure of human α-galactosidase A, determined by X-ray crystallography. α-Galactosidase A is a homodimeric glycoprotein. Each monomer contains five disulphide bonds (Cys52–Cys94, Cys56–Cys63, Cys142–Cys172, Cys202–Cys223 and Cys378–Cys382) and four possible N-glycosylation sites (Asn139, Asn192, Asn215 and Asn408).
Information on the putative three-dimensional structure of α-galactosidase A provides a better insight into how the enzyme works at the molecular level as well as a greater understanding of the molecular pathology of the mutated protein. This is especially the case for missense mutations, which represent the smallest possible structural change of the polypeptide – the replacement of one amino acid by another one. Based on their model, Garman and Garboczi [8] classified GLA missense mutations into three groups. First, mutations that perturb the active site of the enzyme by changing residues that either form the active centre itself or are essential for its correct three-dimensional structure; secondly, buried mutations that affect residues distant from the active site, although they adversely affect the folding and stability of the protein; thirdly, 'other' mutations that do not fall into either of the above categories, although their negative effect on the catabolic function of the molecule is evident, for example by disruption of an important disulphide bond or elimination of an N-carbohydrate attachment site.
By computer modelling of the structure of α-galactosidase A mutants, Matsuzawa and colleagues [9] have provided data suggesting that a number of GLA missense mutations associated with the classic disease phenotype should result in significant structural changes in functionally important regions of the polypeptide and thus in a dysfunctional and unstable enzyme. In contrast, other mutations located distant from the active site should result in small structural changes. Some of these latter enzyme variants, for example p.Met72Val, p.Gln279Glu and p.Met296Ile, which are known to be associated with a mild disease phenotype, had normal Km and Vmax values and showed residual catalytic activity. However, the mutant enzymes were post-translationally inactivated and rapidly degraded. It has been shown that galactose may enhance the stability of the above-mentioned three 'mild' mutants expressed in lymphoblasts.
It is hoped that, eventually, knowledge of structure/function correlates for certain mutations may help to develop new therapies for Fabry disease, such as the use of chaperone molecules (see Chapter 43).
References
- 1.
- Yasuda M, Shabbeer J, Benson SD, Maire I, Burnett RM, Desnick RJ. Fabry disease: characterization of α-galactosidase A double mutations and the D313Y plasma enzyme pseudodeficiency allele. Hum Mutat. 2003;22:486–92. [PubMed: 14635108]
- 2.
- Ishii S, Sakuraba H, Suzuki Y. Point mutations in the upstream region of the α-galactosidase A gene exon 6 in an atypical variant of Fabry disease. Hum Genet. 1992;89:29–32. [PubMed: 1315715]
- 3.
- Altarescu GM, Goldfarb LG, Park KY, Kaneski C, Jeffries N, Litvak S. et al. Identification of fifteen novel mutations and genotype–phenotype relationship in Fabry disease. Clin Genet. 2001;60:46–51. [PubMed: 11531969]
- 4.
- Schaefer E, Mehta A, Gal A. Genotype and phenotype in Fabry disease: analysis of the Fabry Outcome Survey. Acta Paediatr Suppl. 2005;94:87–92. 79. [PubMed: 15895718]
- 5.
- Blanch LC, Meaney C, Morris CP. A sensitive mutation screening strategy for Fabry disease: detection of nine mutations in the α-galactosidase A gene. Hum Mutat. 1996;8:38–43. [PubMed: 8807334]
- 6.
- Shabbeer J, Yasuda M, Luca E, Desnick RJ. Fabry disease: 45 novel mutations in the α-galactosidase A gene causing the classical phenotype. Mol Genet Metab. 2002;76:23–30. [PubMed: 12175777]
- 7.
- Schaefer E, Baron K, Widmer U, Deegan P, Neumann HP, Sunder-Plassmann G. et al. Thirty-four novel mutations of the GLA gene in 121 patients with Fabry disease. Hum Mutat. 2005;25:412. [PubMed: 15776423]
- 8.
- Garman SC, Garboczi DN. The molecular defect leading to Fabry disease: structure of human α-galactosidase. J Mol Biol. 2004;337:319–35. [PubMed: 15003450]
- 9.
- Matsuzawa F, Aikawa SI, Doi H, Okumiya T, Sakuraba H. Fabry disease: correlation between structural changes in α-galactosidase, and clinical and biochemical phenotypes. Hum Genet. 2005;117:317–28. [PubMed: 15924232]
In recent years, several excellent reviews have been published on the GLA gene, its mutations and various aspects of Fabry disease. In order to avoid redundancy, the number of references has been kept to a minimum in this chapter. References to individual mutations are available on request from the authors.
- PubMedLinks to PubMed
- Fabry disease: twenty-three mutations including sense and antisense CpG alterations and identification of a deletional hot-spot in the alpha-galactosidase A gene.[Hum Mol Genet. 1994]Fabry disease: twenty-three mutations including sense and antisense CpG alterations and identification of a deletional hot-spot in the alpha-galactosidase A gene.Eng CM, Niehaus DJ, Enriquez AL, Burgert TS, Ludman MD, Desnick RJ. Hum Mol Genet. 1994 Oct; 3(10):1795-9.
- Alport syndrome. Molecular genetic aspects.[Dan Med Bull. 2009]Alport syndrome. Molecular genetic aspects.Hertz JM. Dan Med Bull. 2009 Aug; 56(3):105-52.
- Thirty-four novel mutations of the GLA gene in 121 patients with Fabry disease.[Hum Mutat. 2005]Thirty-four novel mutations of the GLA gene in 121 patients with Fabry disease.Schäfer E, Baron K, Widmer U, Deegan P, Neumann HP, Sunder-Plassmann G, Johansson JO, Whybra C, Ries M, Pastores GM, et al. Hum Mutat. 2005 Apr; 25(4):412.
- Review Molecular basis of Fabry disease: mutations and polymorphisms in the human alpha-galactosidase A gene.[Hum Mutat. 1994]Review Molecular basis of Fabry disease: mutations and polymorphisms in the human alpha-galactosidase A gene.Eng CM, Desnick RJ. Hum Mutat. 1994; 3(2):103-11.
- Review [alpha-Galactosidase gene mutation and its expression product in Fabry disease (alpha-galactosidase deficiency)].[Rinsho Byori. 1997]Review [alpha-Galactosidase gene mutation and its expression product in Fabry disease (alpha-galactosidase deficiency)].Okumiya T, Takata T, Sasaki M, Sakuraba H. Rinsho Byori. 1997 Feb; 45(2):127-35.
- The genetic basis of Fabry disease - Fabry DiseaseThe genetic basis of Fabry disease - Fabry Disease
Your browsing activity is empty.
Activity recording is turned off.
See more...