Clinical Description
Cleidocranial dysplasia (CCD) spectrum disorder is a skeletal dysplasia representing a clinical continuum ranging from classic CCD (triad of delayed closure of the cranial sutures, hypoplastic or aplastic clavicles, and dental abnormalities), to mild CCD, to isolated dental anomalies without other skeletal features [Golan et al 2000]. Most individuals are diagnosed because they have classic features. CCD spectrum disorder affects most prominently those bones derived from intramembranous ossification, such as the cranium and the clavicles, although bones formed through endochondral ossification can also be affected. Cooper et al [2001] recorded the natural history of 90 probands and 56 first- and second-degree relatives; findings highlight the clinical variability of this condition within affected members of the same family who harbor the same pathogenic variant. Roberts et al [2013] reviewed their experience with more than 100 affected individuals in South Africa.
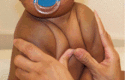
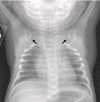
Classic CCD. The most prominent clinical findings in individuals with classic CCD are listed in Suggestive Findings and include: abnormally large, wide-open fontanelles at birth that may remain open throughout life; clavicular hypoplasia resulting in narrow, sloping shoulders that can be opposed at the midline; and abnormal dentition (see Dental complications in this section).
Further medical problems identified in individuals with CCD spectrum disorder include short stature, skeletal/orthopedic findings, dental complications, ENT complications, endocrine findings, and mild developmental delay.
Height. Individuals with CCD spectrum disorder are often shorter than their unaffected sibs and present with postnatal growth deficiency.
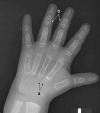
Skeletal/orthopedic findings. Affected individuals are more likely to have other bone-related problems:
Other less common orthopedic problems include joint dislocation at the shoulder and elbow [El-Gharbawy et al 2010].
Dental complications. Up to 94% of persons with CCD spectrum disorder have dental findings, including delayed eruption of secondary dentition and failure to shed the primary teeth [Golan et al 2003]. The most consistent dental findings in individuals with a CCD spectrum disorder are the presence of the second permanent molar with the primary dentition (80%), wide spacing in the lower incisor area, supernumerary tooth germs (70%), and parallel-sided ascending rami [Cooper et al 2001, Golan et al 2003, Golan et al 2004, Bufalino et al 2012]. Individuals with a CCD spectrum disorder are more likely to have an underbite and to have gingival cysts that usually form around extra teeth [McNamara et al 1999].
ENT complications. Recurrent sinus infections and other upper airway complications are observed significantly more often in individuals with CCD spectrum disorder than in the general population. When symptoms are suggestive of upper airway obstruction, a sleep study is indicated, and surgical intervention may be required. Conductive hearing loss occurs in 39% of affected individuals. Individuals with CCD spectrum disorder of any age are more likely to have recurrent ear infections.
Endocrinology. Individuals with CCD spectrum disorder can have low insulin-like growth factor 1 levels. Low vitamin D with no consistent association with osteoporosis has also been reported [Dinçsoy Bir et al 2017]. Rarely, individuals with CCD spectrum disorder have low levels of alkaline phosphatase [Morava et al 2002, Unger et al 2002, El-Gharbawy et al 2010].
Development. Intelligence is typically normal. Children younger than age five years may show mild motor delay, particularly in gross motor abilities. This delay may be associated with orthopedic complications such as flat feet and genu valgum. No significant differences are observed among elementary school-age children.