Summary
Clinical characteristics.
SLC26A2-related multiple epiphyseal dysplasia (SLC26A2-MED) is characterized by joint pain (usually in the hips or knees); malformations of hands, feet, and knees; and scoliosis. Approximately 50% of affected individuals have an abnormal finding at birth, including clubfoot, clinodactyly, or (rarely) cystic ear swelling. Onset of articular pain is variable but usually occurs in late childhood. Stature is usually within the normal range prior to puberty; in adulthood, stature is only slightly diminished and ranges from 150 to 180 cm. Functional disability is mild.
Diagnosis/testing.
Diagnosis of SLC26A2-MED is based on detection of biallelic variants in SLC26A2 by molecular genetic testing in an individual with compatible clinical and radiographic findings.
Management.
Treatment of manifestations: Physiotherapy for muscular strengthening and maintaining mobility; cautious use of analgesic medications such as nonsteroidal anti-inflammatory drugs; orthopedic surgery (joint replacement) as indicated; career counseling.
Prevention of secondary complications: Intensive physiotherapy may help in delaying joint contractures and maintaining mobility.
Surveillance: Radiographs as indicated.
Agents/circumstances to avoid: Sports involving joint overload.
Genetic counseling.
SLC26A2-MED is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for an SLC26A2 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the SLC26A2 pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives and prenatal/preimplantation genetic testing are possible.
Diagnosis
Suggestive Findings
SLC26A2-related multiple epiphyseal dysplasia (SLC26A2-MED) should be suspected in individuals with the following clinical and radiographic features.
Clinical features
Joint pain (usually in the hips and knees). Onset of pain is variable, but usually occurs in late childhood. Some individuals have no pain.
Deformity of hands, feet, and knees
Scoliosis
Radiographic features
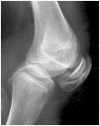
Double-layered patella Ballhausen et al [2003]; reprinted with permission from the BMJ Publishing Group
Family history is consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
The diagnosis of SLC26A2-MED is established in a proband with the characteristic clinical and radiographic features described in Suggestive Findings and biallelic pathogenic (or likely pathogenic) variants in SLC26A2 identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic, and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of biallelic SLC26A2 variants of uncertain significance (or of one known SLC26A2 pathogenic variant and one SLC26A2 variant of uncertain significance) does not establish or rule out the diagnosis of the disorder.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive
genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a phenotype indistinguishable from many other skeletal dysplasias are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
Single-gene testing. Sequence analysis of SLC26A2 detects small intragenic deletions/insertions and missense, nonsense, and splice site variants. Typically, if only one or no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications; however, to date such variants have not been identified as a cause of this disorder.
A multigene panel that includes SLC26A2 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
When the phenotype is indistinguishable from other skeletal dysplasias, comprehensive
genomic testing, which does not require the clinician to determine which gene is likely involved, is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in SLC26A2-Related Multiple Epiphyseal Dysplasia
View in own window
| Gene 1 | Method | Proportion of Pathogenic Variants 2 Detectable by Method |
|---|
|
SLC26A2
| Sequence analysis 3 | 100% 4, 5 |
| Gene-targeted deletion/duplication analysis 6 | None reported 7 |
- 1.
- 2.
- 3.
- 4.
The four most common SLC26A2 pathogenic variants (p.Arg279Trp, c.-26+2T>C, p.Arg178Ter, and p.Cys653Ser) account for approximately 70% of disease alleles in all SLC26A2-related dysplasias. Targeted analysis of these four variants identified at least one pathogenic variant in nearly 100% of individuals with SLC26A2-MED (80% of individuals with SLC26A2-MED have two of the most common pathogenic variants, and another 16% have one common pathogenic variant in compound heterozygosity with another pathogenic variant).
- 5.
- 6.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 7.
Data derived from the subscription-based professional view of Human Gene Mutation Database [Stenson et al 2020]
Clinical Characteristics
Clinical Description
SLC26A2-related multiple epiphyseal dysplasia (SLC26A2-MED) is characterized by early-onset joint pain, malformations of hands, feet, and knees, and scoliosis. Approximately 50% of affected individuals have an abnormal finding at birth, including clubfoot, clinodactyly, cleft palate, or (rarely) cystic ear swelling. However, only half of those with findings at birth are suspected of having a skeletal dysplasia.
Skeletal manifestations. Chronic joint pain most often occurs in the hips, knees, wrists, and fingers. The onset of joint pain is variable; adolescents are usually symptomatic in multiple joints, and joint pain increases after physical exercise.
Waddling gait, hand/foot deformities (mild brachydactyly, clinodactyly, clubfoot, broadening of the space between the first and second toes), and mild scoliosis are also reported. Brachydactyly is evident after puberty in most individuals. Habitus is unremarkable in most affected individuals, except for genu valgum in some.
Stature is usually within the normal range prior to puberty. In adulthood, stature is only slightly diminished, with the median height shifting from the 50th to the tenth centile; range of adult height is 150-180 cm. Approximately one third of affected adults have proportionately short stature that is two standard deviations below the mean for age.
Craniofacial features. Facies are typically normal. Cleft palate may be present. Cystic ear swelling rarely occurs.
Progression. Functional disability is mild or absent in childhood and adolescence [Ballhausen et al 2003]; joint involvement progresses slightly in young adults, but hip and knee surgery is usually not needed. Bowing of the extremities is not observed.
Genotype-Phenotype Correlations
Genotype-phenotype correlations indicate that the amount of residual activity of the sulfate transporter modulates the phenotype in this spectrum of disorders, which extends from lethal achondrogenesis type 1B (ACG1B) to mild SLC26A2-MED. Homozygosity or compound heterozygosity for pathogenic variants predicting stop codons or structural pathogenic variants in transmembrane domains of the sulfate transporter are associated with ACG1B, while pathogenic variants located in extracellular loops, in the cytoplasmic tail of the protein, or in the regulatory 5'-flanking region of the gene result in less severe phenotypes [Superti-Furga et al 1996, Karniski 2001, Maeda et al 2006].
The pathogenic variant p.Arg279Trp is the most common SLC26A2 variant found outside of Finland (45% of alleles); it results in the mild SLC26A2-MED phenotype when homozygous and mostly in diastrophic dysplasia (DTD) and atelosteogenesis type 2 (AO2) phenotypes when found in the compound heterozygous state [Barbosa et al 2011].
Pathogenic variant p.Arg178Ter is the second most common variant (9% of alleles) and is associated with a more severe DTD phenotype or even the perinatal-lethal AO2 phenotype, particularly when combined in trans with the p.Arg279Trp variant.
Pathogenic variants p.Cys653Ser and c.-26+2T>C are the third most common variants (8% of alleles).
Pathogenic variant p.Cys653Ser results in SLC26A2-MED when homozygous and in SLC26A2-MED or DTD when present in trans with other pathogenic variants [Czarny-Ratajczak et al 2010].
Pathogenic variant c.-26+2T>C is sometimes referred to as the "Finnish" variant because it is much more frequent in Finland than in the remainder of the world population. It produces low levels of correctly spliced mRNA and results in DTD when homozygous. It is the only variant that has been identified in all four SLC26A2-related dysplasias, in compound heterozygosity with mild (SLC26A2-MED and DTD) or severe (AO2 and ACG1B) alleles [Dwyer et al 2010].
The same pathogenic variants found in the ACG1B phenotype can also be found in the milder phenotypes (AO2 and DTD) if the second allele is a relatively mild variant. Indeed, missense variants located outside of the transmembrane domain of the sulfate transporter are often associated with residual activity that can "rescue" the effect of a null allele [Rossi & Superti-Furga 2001].
Nomenclature
Multiple epiphyseal dysplasia (MED) is a disorder with clinical and genetic heterogeneity. In the past, the disorder was clinically subdivided into the milder Ribbing type, with flattened epiphysis and normal or near-normal stature; the more severe Fairbank type, with round, small epiphyses and short stature; and the unclassified types [International Working Group on Constitutional Diseases of Bone 1998].
The genetic dissection of this heterogeneous group of conditions in recent years has provided a molecular-pathogenic classification of the different subtypes according to the gene involved:
SLC26A2-related multiple epiphyseal dysplasia (
SLC26A2-MED) is classified in the sulfation disorders group of the 2023 revised Nosology of Genetic Skeletal Disorders [
Unger et al 2023]. It accounts for approximately 25% of cases of MED.
SLC26A2-MED was originally referred to as recessive MED (rMED), and this term continues to be used regularly in the medical literature. However, in order to avoid confusion with the
autosomal recessive CANT1-related
phenotype that is also described as multiple epiphyseal dysplasia (MED) and to move toward dyadic naming, rMED caused by
SLC26A2 pathogenic variants is now designated "multiple epiphyseal dysplasia,
SLC26A2-related" in the 2023 revised nosology [
Unger et al 2023].
The other subtypes of MED are classified in the pseudoachondroplasia and the multiple epiphyseal dysplasias group in the 2023 revised nosology [
Unger et al 2023]. The most frequent form of MED is caused by
autosomal dominant pathogenic variants in
COMP (~50% of cases). The remaining 20%-25% of cases are split between
MATN3,
COL9A1,
COL9A2, and
COL9A3.
Prevalence
Exact data about the prevalence of MED and its subtypes are not available. Based on the number of individuals seen in growth clinics, rheumatology clinics, and genetics clinics, and compared to conditions whose incidences are more precisely known (e.g., achondroplasia, osteogenesis imperfecta), it seems reasonable to estimate an overall prevalence of 1:20,000 [Unger et al 2008]. This prevalence is probably an underestimation, as simplex cases (i.e., a single occurrence in a family) may remain undiagnosed. SLC26A2-MED is one of the most frequent forms of MED, accounting for almost 25% of all individuals diagnosed with MED [Jackson et al 2012].
Differential Diagnosis
SLC26A2-related multiple epiphyseal dysplasia (SLC26A2-MED) needs to be distinguished from the more common autosomal dominant forms of multiple epiphyseal dysplasia (MED). Clinical and radiographic differences between the genetically distinct forms of these skeletal dysplasias may allow clinicians to distinguish between them (see Table 3).
Note: In contrast to autosomal dominant MED, prepubertal children with SLC26A2-MED usually do not have short stature.
Table 3.
Autosomal Dominant Multiple Epiphyseal Dysplasia *
View in own window
| Gene | Clinical Features |
|---|
|
COL9A1
| COL9A1-, COL9A2-, & COL9A3-MED appear to have more severe knee involvement but relative sparing of the hips, resulting in a milder course than MED assoc w/COMP or SLC26A2 pathogenic variants. |
|
COL9A2
|
|
COL9A3
|
|
COMP
| COMP-MED is usually assoc w/significant involvement at the capital femoral epiphyses & irregular acetabula. 1 |
|
MATN3
| MATN3-MED appears to be the mildest form of MED identified to date & is assoc w/a high degree of intrafamilial variability. |
MED = multiple epiphyseal dysplasia
- 1.
CANT1-related MED. Homozygous CANT1 missense variants were reported in four individuals from two families with radiographic phenotypes described as compatible with MED [Balasubramanian et al 2017]. No further individuals have yet been described; thus, given the paucity of available data, no clear clinical and radiographic delineation can be made.
Unclassified
MED. Some individuals with MED do not have pathogenic variants in a known gene [Zankl et al 2007, Unger et al 2008]; in these individuals, MED remains unclassified.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease in an individual diagnosed with SLC26A2-related multiple epiphyseal dysplasia (SLC26A2-MED), the following evaluations (if not performed as part of the evaluation that led to the diagnosis) are recommended:
Height measurement
Elicitation of pain history
Radiographs of the entire spine (AP and lateral), pelvis (AP), and knees (AP and lateral), to determine the extent and severity of joint involvement
Consultation with a medical geneticist, certified genetic counselor, or certified advanced genetic nurse to inform affected individuals and their families about the nature,
mode of inheritance, and implications of
SLC26A2-MED to facilitate medical and personal decision making
Treatment of Manifestations
Symptomatic individuals should be seen by a physical therapist and an orthopedist to assess the possibility of treatment (physiotherapy for muscular strengthening and maintaining mobility, cautious use of analgesic medications such as nonsteroidal anti-inflammatory drugs) and the optimal time for surgery (joint replacement), if indicated.
Intensive physiotherapy may delay joint contractures and help maintain mobility.
Psychosocial support addressing issues of chronic pain and career counseling is warranted.
Surveillance
Radiographic surveillance by an orthopedist is appropriate.
Agents/Circumstances to Avoid
Sports involving joint overload are to be avoided.
Evaluation of Relatives at Risk
Predictive testing of at-risk sibs is not indicated because no preventive measures or therapeutic interventions to reduce morbidity are available.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Women affected by SLC26A2-MED may suffer from chronic joint pain that may increase during pregnancy as a result of maternal weight gain. Appropriate pain management should be offered, and physical therapy should be intensified.
See MotherToBaby for further information on medication use during pregnancy.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
SLC26A2-related multiple epiphyseal dysplasia (SLC26A2-MED) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
Sibs of a proband
Offspring of a proband. Unless an affected individual's reproductive partner also has SLC26A2-MED or is a carrier, offspring will be obligate heterozygotes (carriers) for a pathogenic variant in SLC26A2.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an SLC26A2 pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the SLC26A2 pathogenic variants in the family.
Prenatal Testing and Preimplantation Genetic Testing
Once the SLC26A2 pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and in families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
SLC26A2-Related Multiple Epiphyseal Dysplasia: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Molecular Pathogenesis
SLC26A2 pathogenic variants are responsible for the family of chondrodysplasias including achondrogenesis type 1B (ACG1B), atelosteogenesis type 2 (AO2), diastrophic dysplasia (DTD), and SLC26A2-related multiple epiphyseal dysplasia (SLC26A2-MED). Impaired activity of the sulfate transporter in chondrocytes and fibroblasts results in the synthesis of proteoglycans that are not sulfated or are insufficiently sulfated [Rossi et al 1998, Satoh et al 1998], most likely because of intracellular sulfate depletion [Rossi et al 1996, Gualeni et al 2010]. Undersulfation of proteoglycans affects the composition of the extracellular matrix and leads to impairment of proteoglycan deposition, which is necessary for proper endochondral bone formation [Corsi et al 2001, Forlino et al 2005, Dawson 2011]. The clinical severity can be correlated with the residual activities of the sulfate transporter resulting from different pathogenic variants [Rossi et al 1996, Rossi et al 1997, Corsi et al 2001, Rossi & Superti-Furga 2001, Rossi et al 2003, Karniski 2004, Maeda et al 2006].
In a Xenopus oocyte model, the p.Arg178Ter pathogenic variant was shown to abolish sulfate transporter activity, and the p.Val341del pathogenic variant showed detectable but very low activity (17% of the wild type) of the sulfate transporter [Karniski 2001]. The same variants associated in some individuals with the ACG1B phenotype can be found in individuals with a milder phenotype (AO2 and DTD) if the second allele is a relatively mild variant. Indeed, missense variants located outside the transmembrane domain of the sulfate transporter are often associated with residual activity that can "rescue" the effect of a null allele. Other conclusions from the Xenopus study are at odds with consistent clinical observations, the discrepancy probably being the result of temperature and cellular processing differences between Xenopus oocytes and humans (20 °C vs 37 °C) [Superti-Furga et al 1996, Rossi & Superti-Furga 2001, Superti-Furga 2001, Superti-Furga 2002]. Similar studies conducted in mammalian cells [Karniski 2004] have produced results that are much more consistent with clinical genotype-phenotype correlations. These studies have essentially confirmed predictions that ACG1B-causing variants are associated with no residual transport activity, while the milder phenotypes result from either different combinations of "null" variants with other alleles that allow for some residual activity or from two variants with residual activity. Original observations were: (1) intracellular retention of the sulfate transporter protein with the variant p.Gly678Val; and (2) abnormal molecular weight of the sulfate transporter with p.Gln454Pro, possibly indicating protease sensitivity or aberrant glycosylation.
Mechanism of disease causation. Loss of function
Table 4.
Notable SLC26A2 Pathogenic Variants
View in own window
| Reference Sequences | DNA Nucleotide Change
(Alias 1) | Predicted Protein Change
(Alias 1) | Comment [Reference] |
|---|
|
NM_000112.3
| c.-26+2T>C | -- | Founder variant in Finland; only variant that has been identified in all 4 SLC26A2-related dysplasias, in compound heterozygosity w/mild (SLC26A2-MED & DTD) or severe (AO2 & ACG1B) alleles [Dwyer et al 2010] |
NM_000112.3
NP_000103.2
| c.532C>T
(559C>T) | p.Arg178Ter | Second most common variant (9% of alleles); assoc w/more severe DTD phenotype or perinatal-lethal AO2, esp when combined in trans w/variant p.Arg279Trp |
c.835C>T
(c.862C>T) | p.Arg279Trp | Most common variant found outside of Finland (45% of alleles); mild SLC26A2-MED when homozygous & mostly DTD & AO2 when found in compound heterozygous state [Barbosa et al 2011] |
c.1020_1022delTGT
(1045-1047delGTT) | p.Val341del | See Molecular Pathogenesis. |
c.1361A>C
(1388A>C) | p.Gln454Pro |
c.1957T>A
(1984T>A) | p.Cys653Ser | Third most common variant (8% of alleles); SLC26A2-MED when homozygous & SLC26A2-MED or DTD when present in trans w/other pathogenic variants [Czarny-Ratajczak et al 2010] |
c.2033G>T
(2060G>T) | p.Gly678Val | See Molecular Pathogenesis. |
ACG1B = achondrogenesis type 1B; AO2 = atelosteogenesis type 2; DTD = diastrophic dysplasia; MED = multiple epiphyseal dysplasia
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Variant designation that does not conform to current naming conventions