
Copyright Notice
BLAST is a Registered Trademark of the National Library of Medicine
Bookshelf ID: NBK279684

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
BLAST® Command Line Applications User Manual [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2008-.
The functionality offered by the BLAST+ applications has been organized by program type. The following graph depicts a correspondence between the NCBI C Toolkit BLAST command line applications and the BLAST+ applications:
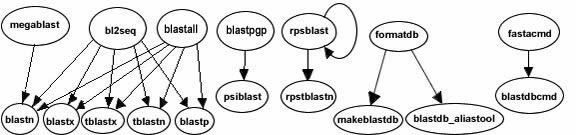
The easiest way to get started using the BLAST+ command line applications is by means of the legacy_blast.pl PERL script which is bundled along with the BLAST+ applications. To utilize this script, simply prefix it to the invocation of the C toolkit BLAST command line application and append the --path option pointing to the installation directory of the BLAST+ applications. For example, instead of using
blastall -i query -d nr -o blast.out
use
legacy_blast.pl blastall -i query -d nr -o blast.out
--path /opt/blast/bin
The purpose of the legacy_blast.pl PERL script is to help users make the transition from the C Toolkit BLAST command line applications to the BLAST+ applications. This script produces its own documentation by invoking it without any arguments.
The legacy_blast.pl script supports two modes of operation, one in which the C Toolkit BLAST command line invocation is converted and executed on behalf of the user and another which solely displays the BLAST+ application equivalent to what was provided, without executing the command.
The first mode of operation is achieved by specifying the C Toolkit BLAST command line application invocation and optionally providing the --path argument after the command line to convert if the installation path for the BLAST+ applications differs from the default (available by invoking the script without arguments). See example in the first section of the Quick start.
The second mode of operation is achieved by specifying the C Toolkit BLAST command line application invocation and appending the --print_only command line option as follows:
$ ./legacy_blast.pl megablast -i query.fsa -d nt -o mb.out --print_only
/opt/ncbi/blast/bin/blastn -query query.fsa -db "nt" -out mb.out
$
All BLAST+ applications have consistent exit codes to signify the exit status of the application. The possible exit codes along with their meaning are detailed in the table below:
|
Exit Code |
Meaning |
|
0 |
Success |
|
1 |
Error in query sequence(s) or BLAST options |
|
2 |
Error in BLAST database |
|
3 |
Error in BLAST engine |
|
4 |
Out of memory |
|
5 |
Network error connecting to NCBI to fetch sequence data |
|
6 |
Error creating output files |
|
255 |
Unknown error |
In the case of BLAST+ database applications, the possible exit codes are 0 (indicating success) and 1 (indicating failure).
This appendix consists of several tables that list option names, types, default values, and a short description of the option. These tables were first published as an appendix to an article in BMC Bioinformatics (BLAST+: architecture and applications). They have been updated for this manual.
Options common to all BLAST+ search applications. An option of type “flag” takes no argument, but if present is true. Some options are valid only for a local search (“remote” option not used), others are valid only for a remote search (“remote” option used).
| option | type | default value | description and notes |
|---|---|---|---|
|
db |
string |
none |
BLAST database name. |
|
query |
string |
stdin |
Query file name. |
|
query_loc |
string |
none |
Location on the query sequence (Format: start-stop) |
|
out |
string |
stdout |
Output file name |
|
evalue |
real |
10.0 |
Expect value (E) for saving hits |
|
subject |
string |
none |
File with subject sequence(s) to search. |
|
subject_loc |
string |
none |
Location on the subject sequence (Format: start-stop). |
|
show_gis |
flag |
N/A |
Show NCBI GIs in report. |
|
num_descriptions |
integer |
500 |
Show one-line descriptions for this number of database sequences. |
|
num_alignments |
integer |
250 |
Show alignments for this number of database sequences. |
|
max_target_seqs |
integer |
500 |
Number of aligned sequences to keep. Use with report formats that do not have separate definition line and alignment sections such as tabular (all outfmt > 4). Not compatible with num_descriptions or num_alignments. Ties are broken by order of sequences in the database. |
|
max_hsps |
integer |
none |
Maximum number of HSPs (alignments) to keep for any single query-subject pair. The HSPs shown will be the best as judged by expect value. This number should be an integer that is one or greater. If this option is not set, BLAST shows all HSPs meeting the expect value criteria. Setting it to one will show only the best HSP for every query-subject pair |
|
html |
flag |
N/A |
Produce HTML output |
|
gilist |
string |
none |
Restrict search of database to GI’s listed in this file. Local searches only. |
|
negative_gilist |
string |
none |
Restrict search of database to everything except the GI’s listed in this file. Local searches only. |
|
entrez_query |
string |
none |
Restrict search with the given Entrez query. Remote searches only. |
|
culling_limit |
integer |
none |
Delete a hit that is enveloped by at least this many higher-scoring hits. |
|
best_hit_overhang |
real |
none |
Best Hit algorithm overhang value (recommended value: 0.1) |
|
best_hit_score_edge |
real |
none |
Best Hit algorithm score edge value (recommended value: 0.1) |
|
dbsize |
integer |
none |
Effective size of the database |
|
searchsp |
integer |
none |
Effective length of the search space |
|
import_search_strategy |
string |
none |
Search strategy file to read. |
|
export_search_strategy |
string |
none |
Record search strategy to this file. |
|
parse_deflines |
flag |
N/A |
Parse query and subject bar delimited sequence identifiers (e.g., gi|129295). |
|
num_threads |
integer |
1 |
Number of threads (CPUs) to use in blast search. |
|
remote |
flag |
N/A |
Execute search on NCBI servers? |
|
outfmt |
string |
0 | alignment view options: 0 = pairwise, 1 = query-anchored showing identities, 2 = query-anchored no identities, 3 = flat query-anchored, show identities, 4 = flat query-anchored, no identities, 5 = XML Blast output, 6 = tabular, 7 = tabular with comment lines, 8 = Text ASN.1, 9 = Binary ASN.1 10 = Comma-separated values 11 = BLAST archive format (ASN.1) 12 = Seqalign (JSON), 13 = Multiple-file BLAST JSON, 14 = Multiple-file BLAST XML2, 15 = Single-file BLAST JSON, 16 = Single-file BLAST XML2, 17 = Sequence Alignment/Map (SAM), 18 = Organism Report Options 6, 7, and 10 can be additionally configured to produce a custom format specified by space delimited format specifiers. The supported format specifiers are: qseqid means Query Seq-id qgi means Query GI qacc means Query accesion sseqid means Subject Seq-id sallseqid means All subject Seq-id(s), separated by a ';' sgi means Subject GI sallgi means All subject GIs sacc means Subject accession sallacc means All subject accessions qstart means Start of alignment in query qend means End of alignment in query sstart means Start of alignment in subject send means End of alignment in subject qseq means Aligned part of query sequence sseq means Aligned part of subject sequence evalue means Expect value bitscore means Bit score score means Raw score length means Alignment length pident means Percentage of identical matches nident means Number of identical matches mismatch means Number of mismatches positive means Number of positive-scoring matches gapopen means Number of gap openings gaps means Total number of gap ppos means Percentage of positive-scoring matches frames means Query and subject frames separated by a '/' qframe means Query frame sframe means Subject frame btop means Blast traceback operations (BTOP) staxids means unique Subject Taxonomy ID(s), separated by a ';'(in numerical order) sscinames means unique Subject Scientific Name(s), separated by a ';' scomnames means unique Subject Common Name(s), separated by a ';' sblastnames means unique Subject Blast Name(s), separated by a ';' (in alphabetical order) sskingdoms means unique Subject Super Kingdom(s), separated by a ';' (in alphabetical order) stitle means Subject Title salltitles means All Subject Title(s), separated by a '<>' sstrand means Subject Strand qcovs means Query Coverage Per Subject (for all HSPs) qcovhsp means Query Coverage Per HSP qcovus is a measure of Query Coverage that counts a position in a subject sequence for this measure only once. The second time the position is aligned to the query is not counted towards this measure. When not provided, the default value is: 'qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore', which is equivalent to the keyword 'std' |
blastn application options. The blastn application searches a nucleotide query against nucleotide subject sequences or a nucleotide database. An option of type “flag” takes no arguments, but if present the argument is true. Four different tasks are supported: 1.) “megablast”, for very similar sequences (e.g, sequencing errors), 2.) “dc-megablast”, typically used for inter-species comparisons, 3.) “blastn”, the traditional program used for inter-species comparisons, 4.) “blastn-short”, optimized for sequences less than 30 nucleotides.
| option | task(s) | type | default value | description and notes |
|---|---|---|---|---|
|
word_size |
megablast |
integer |
28 |
Length of initial exact match. |
|
word_size |
dc-megablast |
integer |
11 |
Number of matching nucleotides in initial match. dc-megablast allows non-consecutive letters to match. |
|
word_size |
blastn |
integer |
11 |
Length of initial exact match. |
|
word_size |
blastn-short |
integer |
7 |
Length of initial exact match. |
|
gapopen |
megablast |
integer |
0 |
Cost to open a gap. See appendix “BLASTN reward/penalty values”. |
|
gapextend |
megablast |
integer |
none |
Cost to extend a gap. This default is a function of reward/penalty value. See appendix “BLASTN reward/penalty values”. |
|
gapopen |
blastn, blastn-short, dc-megablast |
integer |
5 |
Cost to open a gap. See appendix “BLASTN reward/penalty values”. |
|
gapextend |
blastn, blastn-short, dc-megablast |
integer |
2 |
Cost to extend a gap. See appendix “BLASTN reward/penalty values”. |
|
reward |
megablast |
integer |
1 |
Reward for a nucleotide match. |
|
penalty |
megablast |
integer |
-2 |
Penalty for a nucleotide mismatch. |
|
reward |
blastn, dc-megablast |
integer |
2 |
Reward for a nucleotide match. |
|
penalty |
blastn, dc-megablast |
integer |
-3 |
Penalty for a nucleotide mismatch. |
|
reward |
blastn-short |
integer |
1 |
Reward for a nucleotide match. |
|
penalty |
blastn-short |
integer |
-3 |
Penalty for a nucleotide mismatch. |
|
strand |
all |
string |
both |
Query strand(s) to search against database/subject. Choice of both, minus, or plus. |
|
dust |
all |
string |
20 64 1 |
Filter query sequence with dust. |
|
filtering_db |
all |
string |
none |
Mask query using the sequences in this database. |
|
window_masker_taxid |
all |
integer |
none |
Enable WindowMasker filtering using a Taxonomic ID. |
|
window_masker_db |
all |
string |
none |
Enable WindowMasker filtering using this file. |
|
soft_masking |
all |
boolean |
true |
Apply filtering locations as soft masks (i.e., only for finding initial matches). |
|
lcase_masking |
all |
flag |
N/A |
Use lower case filtering in query and subject sequence(s). |
|
db_soft_mask |
all |
integer |
none |
Filtering algorithm ID to apply to the BLAST database as soft mask (i.e., only for finding initial matches). |
|
db_hard_mask |
all |
integer |
none |
Filtering algorithm ID to apply to the BLAST database as hard mask (i.e., sequence is masked for all phases of search). |
|
perc_identity |
all |
integer |
0 |
Percent identity cutoff. |
|
template_type |
dc-megablast |
string |
coding |
Discontiguous MegaBLAST template type. Allowed values are coding, optimal and coding_and_optimal. |
|
template_length |
dc-megablast |
integer |
18 |
Discontiguous MegaBLAST template length. |
|
use_index |
megablast |
boolean |
false |
Use MegaBLAST database index. Indices may be created with the makembindex application. |
|
index_name |
megablast |
string |
none |
MegaBLAST database index name. |
|
xdrop_ungap |
all |
real |
20 |
Heuristic value (in bits) for ungapped extensions. |
|
xdrop_gap |
all |
real |
30 |
Heuristic value (in bits) for preliminary gapped extensions. |
|
xdrop_gap_final |
all |
real |
100 |
Heuristic value (in bits) for final gapped alignment. |
|
no_greedy |
megablast |
flag |
N/A |
Use non-greedy dynamic programming extension. |
|
min_raw_gapped_score |
all |
integer |
none |
Minimum raw gapped score to keep an alignment in the preliminary gapped and trace-back stages. Normally set based upon expect value. |
|
ungapped |
all |
flag |
N/A |
Perform ungapped alignment. |
|
window_size |
dc-megablast |
integer |
40 |
Multiple hits window size, use 0 to specify 1-hit algorithm |
blastp application options. The blastp application searches a protein sequence against protein subject sequences or a protein database. An option of type “flag” takes no arguments, but if present the argument is true. Three different tasks are supported: 1.) “blastp”, for standard protein-protein comparisons, 2.) “blastp-short”, optimized for query sequences shorter than 30 residues, and 3.) “blastp-fast”, a faster version that uses a larger word-size per https://www.ncbi.nlm.nih.gov/pubmed/17921491. This table reflects the 2.2.27 BLAST+ release.
| option | task | type | default value | description and notes |
|---|---|---|---|---|
|
word_size |
blastp |
integer |
3 |
Word size of initial match. Valid word sizes are 2-7. |
|
word_size |
blastp-short |
integer |
2 |
Word size of initial match. |
|
word size |
blastp-fast |
Integer |
6 |
Word size of initial match |
|
gapopen |
blastp |
integer |
11 |
Cost to open a gap. |
|
gapextend |
blastp |
integer |
1 |
Cost to extend a gap. |
|
gapopen |
blastp-short |
integer |
9 |
Cost to open a gap. |
|
gapextend |
blastp-short |
integer |
1 |
Cost to extend a gap. |
|
matrix |
blastp |
string |
BLOSUM62 |
Scoring matrix name. |
|
matrix |
blastp-short |
string |
PAM30 |
Scoring matrix name. |
|
threshold |
blastp |
integer |
11 |
Minimum score to add a word to the BLAST lookup table. |
|
threshold |
blastp-short |
integer |
16 |
Minimum score to add a word to the BLAST lookup table. |
|
Threshold |
Blastp-fast |
Integer |
21 |
Minimum score to add a word to the BLAST lookup table. |
|
comp_based_stats |
Blastp and blastp-fast |
string |
2 | Use composition-based statistics: D or d: default (equivalent to 2) 0 or F or f: no composition-based statistics 1: Composition-based statistics as in NAR 29:2994-3005, 2001 2 or T or t : Composition-based score adjustment as in Bioinformatics 21:902-911, 2005, conditioned on sequence properties 3: Composition-based score adjustment as in Bioinformatics 21:902-911, 2005, unconditionally |
|
comp_based_stats |
blastp-short |
string |
0 | Use composition-based statistics : D or d: default (equivalent to 2) 0 or F or f: no composition-based statistics 1: Composition-based statistics as in NAR 29:2994-3005, 2001 2 or T or t : Composition-based score adjustment as in Bioinformatics 21:902-911, 2005, conditioned on sequence properties 3: Composition-based score adjustment as in Bioinformatics 21:902-911, 2005, unconditionally |
|
seg |
all |
string |
no |
Filter query sequence with SEG (Format: 'yes', 'window locut hicut', or 'no' to disable). |
|
soft_masking |
blastp |
boolean |
false |
Apply filtering locations as soft masks (i.e., only for finding initial matches). |
|
lcase_masking |
all |
flag |
N/A |
Use lower case filtering in query and subject sequence(s). |
|
db_soft_mask |
all |
integer |
none |
Filtering algorithm ID to apply to the BLAST database as soft mask (i.e., only for finding initial matches). |
|
db_hard_mask |
all |
integer |
none |
Filtering algorithm ID to apply to the BLAST database as hard mask (i.e., sequence is masked for all phases of search). |
|
xdrop_gap_final |
all |
real |
25 |
Heuristic value (in bits) for final gapped alignment/ |
|
window_size |
Blastp and blastp-fast |
integer |
40 |
Multiple hits window size, use 0 to specify 1-hit algorithm. |
|
window_size |
blastp-short |
integer |
15 |
Multiple hits window size, use 0 to specify 1-hit algorithm. |
|
use_sw_tback |
all |
flag |
N/A |
Compute locally optimal Smith-Waterman alignments? |
blastx application options. The blastx application translates a nucleotide query and searches it against protein subject sequences or a protein database. Two different tasks are supported: 1.) “blastx” for standard translated nucleotide-protein comparison and 2.) “blastx-fast”, a faster version that uses a larger word-size based on https://www.ncbi.nlm.nih.gov/pubmed/17921491.
| option | task | type | default value | description and notes |
|---|---|---|---|---|
|
word_size |
Blastx |
integer |
3 |
Word size for initial match. Valid word sizes are 2-7. |
|
word_size |
Blastx-fast |
Integer |
6 |
Word size for initial match. |
|
gapopen |
All |
integer |
11 |
Cost to open a gap. |
|
gapextend |
All |
integer |
1 |
Cost to extend a gap. |
|
matrix |
All |
string |
BLOSUM62 |
Scoring matrix name. |
|
threshold |
Blastx |
integer |
12 |
Minimum score to add a word to the BLAST lookup table. |
|
threshold |
Blastx-fast |
Integer |
21 |
Minimum score to add a word to the BLAST lookup table. |
|
seg |
All |
string |
12 2.2 2.5 |
Filter query sequence with SEG (Format: 'yes', 'window locut hicut', or 'no' to disable). |
|
soft_masking |
all |
boolean |
false |
Apply filtering locations as soft masks (i.e., only for finding initial matches). |
|
lcase_masking |
all |
flag |
N/A |
Use lower case filtering in query and subject sequence(s). |
|
db_soft_mask |
all |
integer |
none |
Filtering algorithm ID to apply to the BLAST database as soft mask (i.e., only for finding initial matches). |
|
db_hard_mask |
all |
integer |
none |
Filtering algorithm ID to apply to the BLAST database as hard mask (i.e., sequence is masked for all phases of search). |
|
xdrop_gap_final |
all |
real |
25 |
Heuristic value (in bits) for final gapped alignment. |
|
window_size |
all |
integer |
40 |
Multiple hits window size, use 0 to specify 1-hit algorithm. |
|
strand |
all |
string |
both |
Query strand(s) to search against database/subject. Choice of both, minus, or plus. |
|
query_genetic_code |
all |
integer |
1 |
Genetic code to translate query, see ftp://ftp.ncbi.nih.gov/entrez/misc/data/gc.prt |
|
max_intron_length |
all |
integer |
0 |
Length of the largest intron allowed in a translated nucleotide sequence when linking multiple distinct alignments (a negative value disables linking). |
|
comp_based_stats |
all |
integer |
2 | Use composition-based statistics for blastx: D or d: default (equivalent to 2) 0 or F or f: no composition-based statistics 1: Composition-based statistics as in NAR 29:2994-3005, 2001 2 or T or t : Composition-based score adjustment as in Bioinformatics 21:902-911, 2005, conditioned on sequence properties 3: Composition-based score adjustment as in Bioinformatics 21:902-911, 2005, unconditionally Default = `2' |
tblastn application options. The tblastn application searches a protein query against nucleotide subject sequences or a nucleotide database translated at search time. Two different tasks are supported: 1.) “tblastn” for a standard protein-translated nucleotide comparison and 2.) “tblastn-fast” for a faster version with a larger word-size based on https://www.ncbi.nlm.nih.gov/pubmed/17921491.
| option | task | type | default value | description and notes |
|---|---|---|---|---|
|
word_size |
tblastn |
integer |
3 |
Word size for initial match. Valid word sizes are 2-7. |
|
Word size |
tblastn-fast |
Integer |
6 |
Word size for initial match. |
|
gapopen |
All |
integer |
11 |
Cost to open a gap. |
|
gapextend |
All |
integer |
1 |
Cost to extend a gap. |
|
matrix |
All |
string |
BLOSUM62 |
Scoring matrix name. |
|
threshold |
tblastn |
integer |
13 |
Minimum score to add a word to the BLAST lookup table. |
|
threshold |
tblastn-fast |
Integer |
21 |
Minimum score to add a word to the BLAST lookup table. |
|
seg |
All |
string |
12 2.2 2.5 |
Filter query sequence with SEG (Format: 'yes', 'window locut hicut', or 'no' to disable). |
|
soft_masking |
All |
boolean |
false |
Apply filtering locations as soft masks (i.e., only for finding initial matches). |
|
lcase_masking |
All |
flag |
N/A |
Use lower case filtering in query and subject sequence(s). |
|
db_soft_mask |
All |
integer |
none |
Filtering algorithm ID to apply to the BLAST database as soft mask (i.e., only for finding initial matches). |
|
db_hard_mask |
All |
integer |
none |
Filtering algorithm ID to apply to the BLAST database as hard mask (i.e., sequence is masked for all phases of search). |
|
xdrop_gap_final |
All |
real |
25 |
Heuristic value (in bits) for final gapped alignment. |
|
window_size |
All |
integer |
40 |
Multiple hits window size, use 0 to specify 1-hit algorithm. |
|
db_gen_code |
All |
integer |
1 |
Genetic code to translate subject sequences, see ftp://ftp.ncbi.nih.gov/entrez/misc/data/gc.prt |
|
max_intron_length |
All |
integer |
0 |
Length of the largest intron allowed in a translated nucleotide sequence when linking multiple distinct alignments (a negative value disables linking). |
|
comp_based_stats |
all |
string |
2 | Use composition-based statistics for tblastn: D or d: default (equivalent to 2) 0 or F or f: no composition-based statistics 1: Composition-based statistics as in NAR 29:2994-3005, 2001 2 or T or t : Composition-based score adjustment as in Bioinformatics 21:902-911, 2005, conditioned on sequence properties 3: Composition-based score adjustment as in Bioinformatics 21:902-911, 2005, unconditionally Default = `2' |
tblastx application options. The tblastx application searches a translated nucleotide query against translated nucleotide subject sequences or a translated nucleotide database. An option of type “flag” takes no arguments, but if present the argument is true. This table reflects the 2.2.27 BLAST+ release. Only ungapped searches are supported for tblastx.
| option | type | default value | description and notes |
|---|---|---|---|
|
word_size |
integer |
3 |
Word size for initial match. |
|
matrix |
string |
BLOSUM62 |
Scoring matrix name. |
|
threshold |
integer |
13 |
Minimum word score to add the word to the BLAST lookup table. |
|
seg |
string |
12 2.2 2.5 |
Filter query sequence with SEG (Format: 'yes', 'window locut hicut', or 'no' to disable). |
|
soft_masking |
boolean |
false |
Apply filtering locations as soft masks (i.e., only for finding initial matches). |
|
lcase_masking |
flag |
N/A |
Use lower case filtering in query and subject sequence(s). |
|
db_soft_mask |
integer |
none |
Filtering algorithm ID to apply to the BLAST database as soft mask (i.e., only for finding initial matches). |
|
db_hard_mask |
integer |
none |
Filtering algorithm ID to apply to the BLAST database as hard mask (i.e., sequence is masked for all phases of search). |
|
strand |
string |
both |
Query strand(s) to search against database subject sequences. Choice of both, minus, or plus. |
|
query_genetic_code |
integer |
1 |
Genetic code to translate query, see ftp://ftp.ncbi.nih.gov/entrez/misc/data/gc.prt |
|
db_gen_code |
integer |
1 |
Genetic code to translate subject sequences, see ftp://ftp.ncbi.nih.gov/entrez/misc/data/gc.prt |
|
max_intron_length |
integer |
0 |
Length of the largest intron allowed in a translated nucleotide sequence when linking multiple distinct alignments (a negative value disables linking) |
rpsblast application options. The rpsblast application searches a protein query against the conserved domain database (CDD), which is a set of protein profiles. Many of the common options such as matrix or word threshold are set when the CDD is built and cannot be changed by the rpsblast application. A search ready CDD can be downloaded from ftp://ftp.ncbi.nih.gov/pub/mmdb/cdd/
| Option | Type | Default value | Description and notes |
|---|---|---|---|
| window_size | integer | 40 | Multiple hits window size, use 0 to specify 1-hit algorithm. |
| xdrop_ungap | real | 15 | Heuristic value (in bits) for ungapped extensions |
| xdrop_gap | real | 25 | Heuristic value (in bits) for preliminary gapped extensions. |
| xdrop_gap_final | real | 40 | Heuristic value (in bits) for final gapped alignment. |
| seg | string | 12 2.2 2.5 | Filter query sequence with SEG (Format: 'yes', 'window locut hicut', or 'no' to disable). |
| soft_masking | boolean | false | Apply filtering locations as soft masks (i.e., only for finding initial matches). |
| mt_mode | integer | 0 | Set to 1 if a large number of queries are to be searched and you wish to use multiple threads, as specified by the num_threads argument. |
| comp_based_stats | integer | 2 | Use composition-based statistics for rpsblast: D or d: default (equivalent to 2) 0 or F or f: no composition-based statistics 1: Composition-based statistics as in NAR 29:2994-3005, 2001 2 or T or t : Composition-based score adjustment as in Bioinformatics 21:902-911, 2005, conditioned on sequence properties 3: Composition-based score adjustment as in Bioinformatics 21:902-911, 2005, unconditionally Default = `2' |
Makeblastdb application options. This application builds a BLAST database. An option of type “flag” takes no arguments, but if present the argument is true. Starting with the 2.10.0 release, makeblastdb produces version 5 databases by default, which uses LMDB. LMDB requires virtual memory (at least 600 GB, but 800 GB is recommended) to build an index. If makeblastdb cannot access enough virtual memory, it will produce a message containing the string “mdb_env_open”. Virtual memory is just that (virtual) and doesn’t depend on the hardware in your system. In general, we recommend that BLAST users simply set the virtual memory to unlimited. The other alternative is to use an environment variable (BLASTDB_LMDB_MAP_SIZE) to set the required virtual memory lower, but this runs the risk of LMDB not being able to complete indexing the database. For a smaller database (tens of millions of letters) it may be possible to use a value of 100 million.
| option | type | default value | Description and notes |
|---|---|---|---|
|
in |
string |
stdin |
Input file/database name |
|
input_type |
string |
fasta | Input file type, it may be any of the following: fasta: for FASTA file(s) blastdb: for BLAST database(s) asn1_txt: for Seq-entries in text ASN.1 format asn1_bin: for Seq-entries in binary ASN.1 format |
|
dbtype |
string |
prot |
Molecule type of input, values can be nucl or prot. |
|
title |
string |
none |
Title for BLAST database. If not set, the input file name will be used. |
|
parse_seqids |
flag |
N/A |
Parse bar delimited sequence identifiers (e.g., gi|129295) in FASTA input. |
|
hash_index |
flag |
N/A |
Create index of sequence hash values. |
|
mask_data |
string |
none |
Comma-separated list of input files containing masking data as produced by NCBI masking applications (e.g. dustmasker, segmasker, windowmasker). |
|
out |
string |
input file name |
Name of BLAST database to be created. Input file name is used if none provided. This field is required if input consists of multiple files. |
|
max_file_size |
string |
1GB |
Maximum file size to use for BLAST database. 4GB is the maximum supported by the database structure. |
|
blastdb_version |
integer |
5 |
Version 5 (taxonomy aware) is the default starting with the 2.10.0 release. Value must be 4 or 5. |
|
taxid |
integer |
none |
Taxonomy ID to assign to all sequences. |
|
taxid_map |
string |
none | File with two columns mapping sequence ID to the taxonomy ID. The first column is the sequence ID represented as one of:
The second column should be the NCBI taxonomy ID (e.g., 9606 for human). |
|
metadata_output_prefix |
string |
none |
Path prefix for "files" field in BLASTDB metadata file |
|
logfile |
string |
none |
Program log file (default is stderr). |
Makeprofiledb application options. This application builds an RPS-BLAST database. An option of type “flag” takes no arguments, but if present the argument is true. COBALT (a multiple sequence alignment program) and DELTA-BLAST both use RPS-BLAST searches as part of their processing but use specialized versions of the database. This application can build databases for COBALT, DELTA-BLAST, and a standard RPS-BLAST search. The “dbtype” option (see entry in table) determines which flavor of the database is built.
| option | type | default value | Description and notes |
|---|---|---|---|
|
in |
string |
stdin |
Input file that contains a list of scoremat files (delimited by space, tab, or newline) |
|
binary |
flag |
N/A |
The scoremat files are binary ASN.1 |
|
title |
string |
none |
Title for RPS-BLAST database. If not set, the input file name will be used. |
|
threshold |
real |
9.82 |
Threshold for RPSBLAST lookup table. |
|
out |
string |
input file name |
Name of BLAST database to be created. Input file name is used if none provided. |
|
max_file_size |
string |
1GB |
Maximum file size to use for BLAST database. |
|
dbtype |
string |
rps |
Specifies use for RPSBLAST db. One of rps, cobalt, or delta. |
|
index |
flag |
N/A |
Creates index files. |
|
gapopen |
integer |
none |
Cost to open a gap. Used only if scoremat files do not contain PSSM scores, otherwise ignored. |
|
gapextend |
integer |
none |
Cost to extend a gap by one residue. Used only if scoremat files do not contain PSSM scores, otherwise ignored. |
|
scale |
real |
100 |
PSSM scale factor. |
|
matrix |
string |
BLOSUM62 |
Matrix to use in constructing PSSM. One of BLOSUM45, BLOSUM50, BLOSUM62, BLOSUM80, BLOSUM90, PAM250, PAM30 or PAM70. Used only if scoremat files do not contain PSSM scores, otherwise ignored. |
|
obsr_threshold |
real |
6 |
Exclude domains with maximum number of independent observations below this value (for use in DELTA-BLAST searches). |
|
exclude_invalid |
real |
true |
Exclude domains that do not pass validation test (for use in DELTA-BLAST searches). |
|
logfile |
string |
none |
Program log file (default is stderr). |
Blastdbcmd application options. This application reads a BLAST database and produces reports.
| option | type | default value | description and notes |
|---|---|---|---|
|
db |
string |
nr |
BLAST database name. |
|
dbtype |
string |
guess |
Molecule type stored in BLAST database, one of nucl, prot, or guess. |
|
entry |
string |
none |
Comma-delimited search string(s) of sequence identifiers: e.g.: 555, AC147927, 'gnl|dbname|tag', or 'all' to select all sequences in the database |
|
entry_batch |
string |
none |
Input file for batch processing. The format requires one entry per line; each line should begin with the sequence ID followed by any of the following optional specifiers (in any order): range (format: ‘from-to’, inclusive in 1-offsets), strand (‘plus’ or ‘minus’), or masking algorithm ID (integer value representing the available masking algorithm). Omitting the ending range (e.g.: ‘10-‘) is supported, but there should not be any spaces around the ‘-‘. |
|
pig |
integer |
none |
PIG (protein identity group) to retrieve. |
|
info |
flag |
N/A |
Print BLAST database information. |
|
range |
string |
none |
Range of sequence to extract (Format: start-stop). |
|
strand |
string |
plus |
Strand of nucleotide sequence to extract. Choice of plus or minus. |
|
mask_sequence_with |
string |
none |
Produce lower-case masked FASTA using the algorithm IDs specified. |
|
out |
string |
stdout |
Output file name. |
|
outfmt |
string |
%f | Output format, where the available format specifiers are: %f means sequence in FASTA format %s means sequence data (without defline) %a means accession %g means gi %o means ordinal id (OID) %t means sequence title %l means sequence length %T means taxid %L means common taxonomic name %S means scientific name %P means PIG %mX means sequence masking data, where X is an optional comma-separated list of integers to specify the algorithm ID(s) to display (or all masks if absent or invalid specification). Masking data will be displayed as a series of 'N-M' values separated by ';' or the word 'none' if none are available. For every format except '%f', each line of output will correspond to a sequence. |
|
target_only |
flag |
N/A |
Definition line should contain target GI only. |
|
get_dups |
flag |
N/A |
Retrieve duplicate accessions. |
|
line_length |
integer |
80 |
Line length for output. |
|
ctrl_a |
flag |
N/A |
Use Ctrl-A as the non-redundant definition line separator. |
Makembindex application options. The indexed databases created by makembindex are used by production MegaBLAST software and by a new srsearch utility designed to quickly search for nearly exact matches (up to one mismatch) of short queries against a genomic database. When a FASTA formatted file is used as the input, then masking by lower case letters is incorporated in the index. Makembindex can currently build two types of indices, called “old style” and “new style” indexing. The NCBI offers full support for the new style and has deprecated the old style. A MegaBLAST search with a new style index requires that both the index and the corresponding BLAST database be present. The index structure is described in PMID:18567917. Please cite this paper in any publication that uses makembindex.
| option | type | default value | Description and notes |
|---|---|---|---|
|
input |
string |
stdin |
Input file name or BLAST database name, depending on the value of the iformat parameter. For FASTA formatted input, this parameter is optional and defaults to the program's standard input stream. |
|
output |
string |
none | The resulting index name. The index itself can consist of multiple files, called volumes, called <index_name>.00.idx, <index_name>.01.idx,... This option should not be used with new style indices. |
|
iformat |
string |
fasta |
The input format selector. Possible values are 'fasta' and 'blastdb'. |
|
old_style_index |
boolean |
false |
The old_style_index is no longer supported. If set to 'false' the new style index is created. New style indices require a BLAST database as input (use -iformat blastdb), which can be downloaded from the NCBI FTP site or created with makeblastdb. The option -output is ignored for a new style index. New style indices are always created at the same location as the corresponding BLAST database. |
|
db_mask |
integer |
None |
Exclude masked regions of BLAST db from the index. Use makeblastdb to discover the algorithm ID to be used as input for this argument. |
|
legacy |
boolean |
true |
This is a compatibility feature to support current production MegaBLAST. If true, then -stride, -nmer, and -ws_hint are ignored. The legacy format must be used for BLAST. |
|
nmer |
integer |
12 |
N-mer size to use. Ignored if –legacy is specified |
|
ws_hint |
integer |
28 |
This is an optimization hint for makembindex that indicates an expected minimum match size in searches that use the index. If n is the value of -nmer parameter and s is the value of –stride parameter, then the value of -ws_hint must be at least n + s - 1. |
|
stride |
integer |
5 |
makembindex will index every stride-th N-mer of the database. |
|
volsize |
integer |
1536 |
Target index volume size in megabytes. |
BLASTN uses a simple approach to score alignments, with identically matching bases assigned a reward and mismatching bases assigned a penalty. It is important to choose reward/penalty values appropriate to the sequences being aligned with the (absolute) reward/penalty ratio increasing for more divergent sequences. A ratio of 0.33 (1/-3) is appropriate for sequences that are about 99% conserved; a ratio of 0.5 (1/-2) is best for sequences that are 95% conserved; a ratio of about one (1/-1) is best for sequences that are 75% conserved [2].
For each reward/penalty pair, a number of different gap costs are supported. A gap cost includes a value to open the gap and a value to extend the gap by a base. Following the convention of the command-line applications, these costs are listed as positive numbers here. MegaBLAST uses a specialized algorithm to calculate the default gap costs for a reward/penalty pair that is described in PMID:10890397. Briefly, the default megaBLAST cost to open a gap is zero and the cost to extend a gap two letters is given by the absolute value of two mismatches minus one match. For example, given a reward of 1 and penalty of -5, the cost to extend a gap by one letter is 5.5. The default gap costs for other tasks supported by the blastn application is 5 to open a gap and 2 to extend one base.
Table D1 presents the supported reward/penalty values and gap costs.
Supported reward/penalty values and gap costs for the blastn application. The left-most column presents the supported reward/penalty values. The middle column presents pairs of numbers for the cost to open and extend a gap for each reward/penalty value. Blastn also supports gap costs more stringent than those listed (e.g., for reward/penalty of 1/-3 gap costs of 5/2 or 500/2 are supported). The reward/penalty values are ordered from most to least stringent, with the more stringent values better suited for alignments with high sequence identity. The default megaBLAST gap costs are shown in the right-most column. Accurate statistics for these default megaBLAST gap costs can only be calculated for the most stringent reward/penalty values, but the values listed in the middle column can always be used.
| reward/penalty | gap costs (open/extend) | default MegaBLAST gap costs (open/extend) |
|---|---|---|
| 1/-5 | 3/3 | 0/5.5 |
| 1/-4 | 1/2, 0/2, 2/1, 1/1 | 0/4.5 |
| 2/-7 | 2/4, 0/4, 4/2, 2/2 | 0/8 |
| 1/-3 | 2/2, 1/2, 0/2, 2/1, 1/1 | 0/3.5 |
| 2/-5 | 2/4, 0/4, 4/2, 2/2 | 0/6 |
| 1/-2 | 2/2, 1/2, 0/2, 3/1, 2/1, 1/1 | 0/2.5 |
| 2/-3 | 4/4, 2/4, 0/4, 3/3, 6/2, 5/2, 4/2, 2/2 | 0/4 |
| 3/-4 | 6/3, 5/3, 4/3, 6/2, 5/2, 4/2 | N/A |
| 4/-5 | 6/5, 5/5, 4/5, 3/5 | N/A |
| 1/-1 | 3/2, 2/2, 1/2, 0/2, 4/1, 3/1, 2/1 | N/A |
| 3/-2 | 5/5 | N/A |
| 5/-4 | 10/6, 8/6 | N/A |
BLAST uses a substitution matrix for any program that aligns residues. The program may align residues because both the query and database consist of proteins (e.g. BLASTP) or the program may align DNA translated to protein with protein (e.g. BLASTX). A key element in evaluating the quality of a pairwise sequence alignment is the "substitution matrix", which assigns a score for aligning any possible pair of residues. The theory of amino acid substitution matrices is described in [1], and applied to DNA sequence comparison in [2]. In general, different substitution matrices are tailored to detecting similarities among sequences that are diverged by differing degrees [1-3]. A single matrix may nevertheless be reasonably efficient over a relatively broad range of evolutionary change [1-3]. Experimentation has shown that the BLOSUM-62 matrix [4] is among the best for detecting most weak protein similarities. For particularly long and weak alignments, the BLOSUM-45 matrix may prove superior. A detailed statistical theory for gapped alignments has not been developed, and the best gap costs to use with a given substitution matrix are determined empirically. Short alignments need to be relatively strong (i.e. have a higher percentage of matching residues) to rise above background noise. Such short but strong alignments are more easily detected using a matrix with a higher "relative entropy" [1] than that of BLOSUM-62. In particular, short query sequences can only produce short alignments, and therefore database searches with short queries should use an appropriately tailored matrix. The BLOSUM series does not include any matrices with relative entropies suitable for the shortest queries, so the older PAM matrices [5,6] may be used instead. For proteins, a provisional table of recommended substitution matrices and gap costs for various query lengths is:
| Query Length | Substitution Matrix | Gap Costs |
|---|---|---|
| <35 | PAM-30 | (9, 1) |
| 35-50 | PAM-70 | (10, 1) |
| 50-85 | BLOSUM-80 | (10, 1) |
| >85 | BLOSUM-62 | (11, 1) |
Gap Costs
The raw score of an alignment is the sum of the scores for aligning pairs of residues and the scores for gaps. Gapped BLAST and PSI-BLAST use "affine gap costs" which charge the score -a for the existence of a gap, and the score -b for each residue in the gap. Thus a gap of k residues receives a total score of -(a+bk); specifically, a gap of length 1 receives the score -(a+b).
Lambda Ratio
To convert a raw score S into a normalized score S' expressed in bits, one uses the formula S' = (lambda*S - ln K)/(ln 2), where lambda and K are parameters dependent upon the scoring system (substitution matrix and gap costs) employed [7-9]. For determining S', the more important of these parameters is lambda. The "lambda ratio" quoted here is the ratio of the lambda for the given scoring system to that for one using the same substitution scores, but with infinite gap costs [8]. This ratio indicates what proportion of information in an ungapped alignment must be sacrificed in the hope of improving its score through extension using gaps. We have found empirically that the most effective gap costs tend to be those with lambda ratios in the range 0.8 to 0.9.
Introduction
BLAST performs several steps as it searches through a database and winnows the matches, finding the most significant matches that it finally presents to the user. The initial step in this process is the fastest and examines every sequence. Each successive step takes longer but examines fewer sequences. The outline below provides details on the process and a figure provides a visual representation. This outline applies only to gapped BLAST. A letter and number in the figure (e.g., C3) refers to a step in the outline. BLAST is described in greater detail in https://www.ncbi.nlm.nih.gov/pubmed/9254694.
Outline
Read in user query and preprocess (mask for low-complexity, etc.)
Read user options and set parameters for the search. This includes examining how many matches (database sequences) the user wants returned and the expect value. If the user wants N database sequences returned and sets an expect value of E, then:
For Composition-based statistics (CBS), set an (internal) maximum limit of N_i=2*N+50 database sequences and an internal expect value of E_i = 5*E. CBS applies only to protein-protein comparisons and is available for BLASTP, BLASTX, TBLASTN, RPSBLAST, and RPSTBLASTN.
Otherwise, set a maximum limit of N_i=MAX(MIN(2*N, N+50),10) database sequences.
Loop over every sequence in the database, performing the following actions:
Scan for initial matching word hits. If an initial hit is found, then move on to step 2, otherwise move on to next sequence. Example initial matching word hits are:
11 bases exact match for BLASTN.
28 bases exact match for MegaBLAST
3 residue match with score above threshold for BLAST[PX]/TBLASTN (default requires 2 word hits on a diagonal)
6 residue match with score above threshold for BLAST[PX]/TBLASTN for fast task “blastp-fast” etc. (default requires 2 on diagonal)
Perform a gap free extension based on the initial word hits. If this extension has a score above S_g (set so that about one in 50 database sequences pass) then move on to step 3. Otherwise move on to next sequence.
Perform a gapped extension based on the gap free extension. This gapped extension does not collect traceback information, but only the extent of the alignment and the resulting score (making it fast). This gapped extension uses a modified dynamic programming algorithm that only explores a limited space based on a parameter called X_g. If the resulting alignment passes the score cutoff (determined by expect value) move on to next step, otherwise move on to next sequence.
Save the result for further processing unless there are already N_i better matching sequences saved. Save the results in order of significance, keeping the best N_i thus far. Move on to next sequence.
For each entry in the list saved in step C4 above:
Perform a gapped alignment with traceback (i.e., collect score, extent, position of indels, etc.) using an X_fg that is larger than X_g. The larger X_fg means that the score and ranking of a match may change. If CBS, then also adjust the score and expect value based on the composition of the subject sequence (the composition of the query is always considered). This may change the score and ranking of a match, sometimes dramatically.
Add the resulting match to a new ordered list. A tie (two matches with identical score and expect value) is broken by the order of the sequences in the database. Almost every entry processed in the last step results in a significant match, but an alignment calculated with CBS may become much less statistically significant and will no longer be saved.
Format a report based on the list saved in D2:
Discard the N_i-N least significant matches.
Print results for the first N matches.
The retention of N_i > N matches through the internals of BLAST is intended to ensure that if some matches become more or less significant, in the last phase of constructing the alignment, that BLAST will still show the user the most relevant matches. For CBS, we increase N_i by a larger amount than for standard gapped BLAST, as the application of CBS may result in a larger change in the significance of a match. For the same reason, the internal expect value is also increased from the user requested value if CBS is requested.
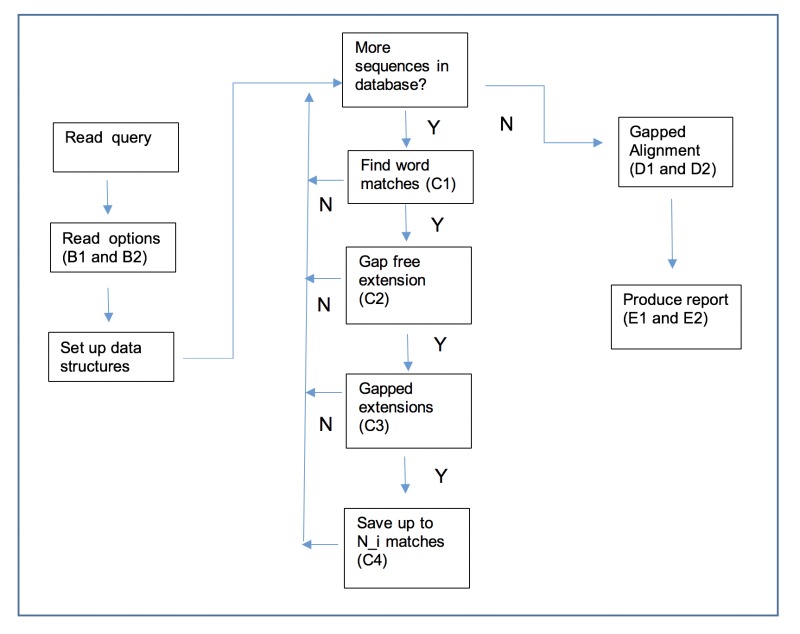
Outline of the BLAST process. A letter and number (e.g., C3) refers to a step in the outline.
BLAST is a Registered Trademark of the National Library of Medicine
Your browsing activity is empty.
Activity recording is turned off.
See more...