Clinical Description
Infantile neuroaxonal dystrophy
(INAD). Onset of INAD usually occurs between ages six months and three years. The disease presents with psychomotor regression (i.e., loss of previously acquired milestones) or delay, delayed walking, or gait disturbance. A single individual with neonatal onset has been reported, with severe hypotonia and marked weakness [Fusco et al 2015].
Truncal hypotonia is observed early in the disease course. Over time, affected persons develop a spastic tetraparesis, with symmetric pyramidal tract signs on clinical examination.
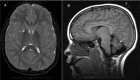
Visual signs and symptoms are common. Strabismus and nystagmus are early features of the disease. Later optic atrophy occurs in most individuals. Optic atrophy may be observed early as optic nerve pallor; thin optic chiasm and tracts have also been reported on brain MRI [Farina et al 1999].
Seizures occur in a minority of individuals as a later symptom [Nardocci et al 1999, Wu et al 2009].
Autonomic involvement may present early as constipation or cold extremities. With progression, some individuals require body temperature monitors because of dangerous fluctuations in core body temperature.
The progression of disease is usually rapid. Many affected children never learn to walk or lose this ability shortly after attaining it. During the end stages of disease, severe spasticity, progressive cognitive decline, and visual impairment result in a vegetative state. Death occurs as a result of secondary illnesses such as aspiration pneumonia, associated with bulbar dysfunction. Many affected children do not survive beyond their first decade, but some survive into their teens or later. Supportive care can contribute to a longer life span by reducing the risk of infection and other complications.
Atypical NAD. Whereas the features of INAD are relatively homogeneous, atypical disease is quite varied.
In general, onset in atypical NAD is in early childhood but can be as late as the late teens. In a series of 13 individuals, four had onset by age three years but a fairly stable course during early childhood resembling static encephalopathy, followed by neurologic deterioration between ages seven and 12 years [Nardocci et al 1999].
The presenting signs and symptoms may be similar to INAD, including gait instability or ataxia. Others may present with speech delay and autistic features, which may remain as the only evidence of disease for a year or more, given the slow progression of atypical NAD compared to INAD [Gregory et al 2008].
Although spastic tetraparesis is evident late in the disease, it is rarely preceded by early truncal hypotonia. In contrast to classic disease, extrapyramidal findings (i.e., dystonia and dysarthria) predominate in atypical NAD. Eye findings are similar to those seen in classic INAD. Neuropsychiatric disturbances including impulsivity, poor attention span, hyperactivity, and emotional lability are also common [Gregory et al 2008].
Atypical NAD is rare, and the life span is not known; however, it is expected to be longer than that observed in classic disease.
PLA2G6-related dystonia-parkinsonism. To date, only a small number of affected individuals have been described [Karkheiran et al 2015]. Age at onset has varied from four to 37 years [Paisán-Ruiz et al 2009, Paisán-Ruiz et al 2010, Yoshino et al 2010, Bower et al 2011, Paisán-Ruiz et al 2012, Virmani et al 2014]; however, the majority have presented in early adulthood (late teens to 20s). Of those with childhood onset, one presented with foot drag and dystonia at age ten years and the other two children presented with an unsteady gait at ages six and eight years. The youngest individual presented with stuttering speech, clumsiness, and dyslexia at age four years – findings that may not be related to the PLA2G6-associated neurodegeneration (PLAN). In young adults, initial symptoms are frequently neuropsychiatric, including depression, personality changes, aggression, delusions, or paranoia. Gait disturbance is also common at presentation.
Regardless of the age at onset, affected individuals consistently develop dystonia and parkinsonism (which may be accompanied by rapid cognitive decline) in their late teens to early twenties. Neuropsychiatric changes may precede the movement disorder or occur concomitantly. Dystonia is most common in the hands and feet but may be more generalized. The most common features of parkinsonism in these individuals are bradykinesia, resting tremor, rigidity, and postural instability. Of note, it is common to have an initially dramatic positive response to dopaminergic agents; however, this tends to be short-lived and followed quickly by the development of motor fluctuations and dyskinesias.
Neuropathology.
Paisán-Ruiz et al [2012] described the neuropathologic findings in seven individuals who spanned the three forms of PLAN. Numerous axonal swellings in the basal ganglia and brain stem were observed in individuals with infantile-onset and adult-onset PLAN. They were also found in the spinal cord in the two individuals for whom cord tissue was available. Lewy bodies were widespread in both those with adult-onset and those with infantile-onset PLAN. In two affected individuals, one with onset at 18 years and the other only specified as "childhood," the Lewy body pathology was comparable to that seen in severe, end-stage Parkinson disease. Tau pathology, to varying degrees, was also found across the PLAN spectrum.