

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Van Dongen AM, editor. Biology of the NMDA Receptor. Boca Raton (FL): CRC Press/Taylor & Francis; 2009.

13.1. INTRODUCTION
NMDA receptors (NMDARs) are glutamate-gated cation channels with high calcium permeability that play important roles in many aspects of the biology of higher organisms. They are critical for the development of the central nervous system (CNS), generation of rhythms for breathing and locomotion, and the processes underlying learning, memory, and neuroplasticity. Consequently, abnormal expression levels and altered NMDAR function have been implicated in numerous neurological disorders and pathological conditions. NMDAR hypofunction can result in cognitive defects, whereas overstimulation causes excitotoxicity and subsequent neurodegeneration. Therefore, NMDARs are important therapeutic targets for many CNS disorders [1–8] including stroke, hypoxia, ischemia, head trauma, Huntington’s, Parkinson’s, and Alzheimer’s diseases, epilepsy, neuropathic pain, alcoholism, schizophrenia, and mood disorders. To date, drugs targeting NMDARs have had only limited success clinically due to poor efficacy and unacceptable side effects, including hallucinations, catatonia, ataxia, nightmares, and memory deficits.
A detailed understanding of the mechanisms underlying agonist-induced receptor activation would facilitate development of more selective drugs that target specific NMDAR subtypes and alter their function to a well-defined extent. This chapter will investigate the physiological roles NMDARs play in the mammalian nervous system and the molecular and structural basis of NMDAR activation. One of the main questions that will be addressed is how agonist binding results in opening of the NMDAR ion channel. Although the mechanism coupling ligand binding to channel opening remains incompletely understood for NMDARs, we propose that this process suggests promising approaches to drug design.
13.2. FUNCTION OF NMDA RECEPTORS
NMDARs belong to a class of ionotropic glutamate receptors (iGluRs) that also includes the AMPA receptors (AMPARs) and kainate receptors [9]. The names of these subclasses derived from the selective synthetic agonists that can be used to distinguish them (see Chapter 12 for detailed description of NMDAR pharmacology). This pharmacological distinction is mirrored by distinct neurophysiological roles for each of the iGluR subtypes.
13.2.1. Synaptic Function
Excitatory synaptic transmission in the vertebrate brain relies on the release of L-glutamate from presynaptic terminals that diffuses across the synaptic cleft and binds to postsynaptic AMPARs and NMDARs. Activation of AMPARs is fast and transient, causing brief depolarizations that last no longer than a few milliseconds. NMDARs are not critical for this basal synaptic transmission, but instead they regulate functional and structural plasticity of individual synapses, dendrites, and neurons by allowing activation of specific calcium-dependent signaling cascades. Several unique properties of NMDARs prevent their activation by L-glutamate released during a single synaptic event.
First, NMDARs activate significantly slower than AMPARs and kainate receptors. Glutamate released from a presynaptic terminal following arrival of an action potential is removed efficiently from the synaptic cleft by the actions of glutamate transporters located in the presynaptic terminal and nearby astrocytes [10,11]. Consequently, glutamate is available for receptor binding only briefly during low frequency synaptic transmission. Because NMDARs have relatively a high affinity for glutamate, the millisecond-long neurotransmitter pulses should be able to partially (and slowly) activate NMDARs. However, individual excitatory synaptic inputs received during baseline activity do not result in calcium (Ca2+) influx because of a second NMDAR property: its pronounced voltage dependence.
At resting membrane potentials, external magnesium (Mg2+) ions enter the NMDAR pore, but unlike the permeant Ca2+ ions, they bind tightly and prevent further ion permeation [12,13]. Mg2+ ions are present at millimolar concentrations in the external milieu of neurons, while intracellular Mg2+ concentrations are in the micromolar range, resulting in a net inward driving force for Mg2+ ions at negative membrane potentials. A depolarization of sufficient amplitude and duration is required to dislodge and repel the Mg2+ ions from the pore, thereby allowing the flow of permeant ions. As a result, the NMDAR acts as a molecular coincidence detector [14]: efficient activation and ion permeation through the NMDAR requires both a sufficiently strong depolarization and synaptic release of glutamate. This dual input requirement, together with their slow activation and deactivation kinetics allows NMDARs to integrate and decode incoming synaptic activity. The high Ca2+ permeability of NMDARs enables them to transduce specific synaptic input patterns into long-lasting alterations in synaptic strength.
13.2.2. Long-Term Potentiation and Depression
The strong depolarization required to remove Mg block from synaptically localized NMDARs can be achieved in several ways. High frequency synaptic inputs may allow the excitatory postsynaptic potentials (EPSPs) generated by AMPAR activation to accumulate and build over time. This phenomenon underlies the paradigm of long-term potentiation (LTP) discovered in 1973 by Bliss and Lomo [15] in which a short burst of high frequency synaptic input (15 Hz for 15 sec or 100 Hz for 3 sec) results in strengthening of excitatory synapses for a prolonged period (hours to days).
The role of NMDARs in LTP induction in the hippocampal CA1 area is well documented [16]. LTP is NMDAR-dependent in many other regions of the brain [17], although NMDAR-independent LTP has also been observed [18,19]. The synaptic strengthening observed during LTP has been attributed to two major mechanisms: (1) phosphorylation of AMPARs, resulting in an increased open probability and (2) enhanced trafficking of AMPARs to the postsynaptic membrane (see Chapter 8 for details).
Long-term depression (LTD), the counterpart of LTP, may be experimentally induced by prolonged low frequency (0.5 to 3 Hz) stimulation of excitatory synapses [20]. Induction of LTD is also NMDAR-dependent in the hippocampal CA1 region and, like LTP induction, requires Ca2+ influx through NMDARs [21]. LTD can also be induced by other mechanisms including stimulation of metabotropic glutamate receptors [22]. The mechanism by which NMDA-dependent LTD reduces synaptic strength is to reverse the effects of LTP: dephosphorylation of AMPARs, thus reducing their open probability [23] and removal of AMPARs from the synaptic plasma membrane by endocytosis (see Chapter 8). In these examples of homosynaptic LTP and LTD, the synapse that receives the low/high frequency input is weakened/strengthened. However, synaptic strength can also be altered in either direction if a single synaptic input is coupled with a postsynaptic depolarization, resulting in heterosynaptic LTP, which has been proposed as a model for associative memory. Postsynaptic depolarizations can occur by various mechanisms. In many cases, neuronal dendrites play critical roles in the generation and processing of these plasticity-inducing signals.
13.2.3. Dendritic Function
Dendrites exhibit active conductances, mediated by voltage-gated Na and Ca channels, as well as the NMDAR itself, which allow them to generate a back-propagating action potential (bAP) [24,25]. Dendritic bAPs have a longer duration than axonal spikes and therefore permit the removal of Mg2+ block from NMDARs. Any glutamate release occurring during a bAP-induced depolarization may therefore result in Ca2+ influx through activated NMDARs and subsequent alterations in synapse strength [26,27]. Because bAPs take time to propagate down distal dendrites, efficient activation of NMDARs by this mechanism requires precise timing of synaptic input relative to bAP generation.
The direction and magnitude of the resulting alteration in synaptic strength depend critically on the temporal relationship between the two processes [28]. The precise timing dependence of presynaptic firing relative to the firing of the postsynaptic neuron, has given rise to a model of long-term synaptic plasticity called spike timing-dependent plasticity (STDP) [29–35]. This promising model extends the LTP–LTD paradigm by proposing that the coupling strength between neurons depends on the degree of correlation of their spiking activities [36].
Thus, STDP implements an important and long ignored aspect of the most influential model for synaptic strengthening formulated in 1949 by Donald Hebb [37]. In Hebb’s words (relevant wording in bold): “When an axon of cell A is near enough to excite a cell B and repeatedly or persistently takes part in firing it, some growth process or metabolic change takes place in one or both cells such that A’s efficiency, as one of the cells firing B, is increased.” Hebb’s insistence that the presynaptic neuron causes the postsynaptic neuron to fire an action potential appears visionary.
The voltage dependence and ion selectivity of NMDARs provide them with regenerative properties that allow the generation of dendritic action potentials (NMDA spikes) that do not originate from bAPs, but from highly synchronized excitatory synaptic inputs to a region of the dendrite [38–42] (see Section 9.4). The ability to generate NMDA spikes endows dendrites with interesting nonlinear computational capabilities, the impact of which is only now beginning to become clear [43–45].
13.2.4. Privileged Ca2+ Ions
The long-lasting effects on synaptic efficacy resulting from activation of NMDARs depend on Ca2+ influx into the postsynaptic compartment (see Chapter 9 for a review of NMDAR-mediated Ca signaling in dendritic spines). It is intriguing that both synaptic strengthening and weakening are mediated by Ca2+ influx through NMDARs. Several hypotheses have been proposed to explain this phenomenon. The magnitude of the rise in Ca2+ concentration may be an important determinant. The direction of change in synaptic strength may also be determined by the temporal properties of the internal Ca concentration changes [46].
Alternatively, there may be a spatial difference in calcium rises associated with by LTP and LTD. NR2B receptors have been proposed to be localized extrasyn-aptically [47] (although this is not a universal finding [48]). Such spatial separation of NR2A and NR2B receptors would allow Ca2+ ions to activate distinct signaling complexes [49,50]. An earlier proposal that the NR2 subtype determines the direction of synaptic change, with activation of NR2A- and NR2B-containing NMDARs resulting in LTP and LTD, respectively [51,52], has not held up in later experiments [53,54]. More work is needed to define the molecular mechanism by which spatiotemporal differences in Ca concentrations resulting from activation of synaptic and extrasynaptic NMDARs of varying subtype compositions lead to bidirectional, long-term changes in synaptic efficacy.
Ca2+ ions entering the neuron through the NMDAR are privileged, because they are able to act locally on large signal transduction complexes associated with synaptic NMDARs, which consist of calcium-dependent enzymes, second messengers, protein kinases and phosphatases, scaffolding proteins, cytoskeletal elements, GTP binding proteins and their regulators, and adhesion molecules [55–57]. NMDAR-mediated calcium influx activates these complexes, generating signal transduction cascades that produce long-lasting changes in synaptic function and structure [57–63].
The ability of an NMDAR to integrate and decode synaptic inputs and generate or amplify dendritic spikes depends on its kinetics of activation, deactivation, and desensitization. The amount of Ca influx is also largely determined by kinetics, in particular the rate of deactivation. Subunit composition is an important determinant for NMDAR (de-)activation kinetics and intracellular calcium dynamics, as discussed in Chapter 9. The amount of Ca influx resulting from NMDAR activation is further modulated by other external physiological factors including pH, zinc ions, and polyamines that are discussed in Chapters 11 and 12. The next section considers the roles of the two coagonists, glutamate and glycine, in activation of NMDARs.
13.2.5. Coagonists Glutamate and Glycine
NMDARs are unique among ligand-gated ion channels in that their activation requires binding of two coagonists, glycine and L-glutamate [64–66]. Glycine is sometimes cited in the literature as an NMDAR modulator—to set it apart from the agonist L-glutamate, but as explained below, their binding sites are structurally similar and seem to play equivalent roles in receptor activation. Physiologically, however, glycine and glutamate have distinct functions. While L-glutamate is released from specific presynaptic terminals, low concentrations of ambient glycine present at the synapse are thought to be sufficient to allow receptor activation. Interestingly, a recent paper suggests that D-serine released by astrocytes is the endogenous glycine site agonist in certain brain regions, allowing glial cells to actively control synaptic metaplasticity [67].
Because glycine plays a more modulatory role in vivo [68,64], while glutamate is the ‘active’, released neurotransmitter, the glycine and glutamate binding sites on the NMDAR represent two distinct therapeutic targets. Particular efforts have been devoted to the development of partial agonists for the glycine site [69–76] that may act as negative modulators of NMDAR function by competitively displacing the full agonist glycine from its binding site. The pharmacology of NMDARs is described in detail in Chapter 12.
An interesting but poorly studied property of glycine is its ability to partially activate NMDARs in the absence of glutamate. This action has been observed in NMDARs containing NR1 and NR2A subunits expressed in Xenopus laevis oocytes, where 10 μM glycine alone activated the receptor by a few percent [77]. Due to the inhibition of this effect by the competitive glutamate-site antagonist APV, it has been suggested that glycine may act as a partial agonist at the glutamate binding site [77]. It is not clear whether ambient glycine in the CNS also exhibits this property, which would allow a small amount of Ca influx through NMDARs independent of synaptic input during large depolarizations that remove Mg2+ block. Also, it is not known whether D-serine, which can be released from astrocytes, has the same ability as glycine to partially activate NMDARs in the absence of glutamate.
The importance of ambient glycine binding to NMDARs for cognitive function is underscored by experiments with transgenic mice carrying mutant alleles of the NR1 subunit [78]. Mice that express NR1 subunits with lowered glycine affinity display a spectrum of cognitive and learning defects including nonhabituating hyperactivity, increased stereotyped behavior, disruptions of nest building activity, and poor performance in the Morris water maze [78], a measure of cued learning. The behavioral phenotypes of these glycine-insensitive mutant mice resemble some of the positive and negative symptoms displayed by schizophrenia patients, consistent with NMDAR hypofunction as one of the leading hypotheses for schizophrenia [79–82]. Section 13.3.7 discusses the (limited) successes of glycine site partial agonists in the clinic. The next two sections describe studies of the behavior of individual NMDAR channels.
13.2.5. Single Channel Behavior
Ion channels are unique among proteins in that their behavior can be studied at the level of a single molecule. This important experimental paradigm was made possible by the advent of the patch clamp technique pioneered by Neher and Sakmann in the mid 1970s [83]. In single channel recordings, individual ion channels are seen to alternate stochastically between two states, open and closed. No measurable ion flux is present in the closed state whereas the open state is characterized by a constant, channel-specific conductance. Activation of an ion channel results from an increase in the probability of being in the open state, not a change in its single channel conductance.
Analysis of single channel behavior provides detailed information about mechanisms of action that cannot be extracted from macroscopic currents measured in whole cell recordings [84]. Early single channel studies suggested that Mg2+ ions inhibited NMDARs via an open channel block mechanism, as indicated by the presence of short closings within otherwise stable open periods [12]. The allosteric modulator spermine exerts multiple effects on single channel behavior, increasing opening frequency at low concentrations, while decreasing single channel conductance and mean open time at higher concentrations, suggesting the existence of two binding sites [85]. Single channel analysis of ethanol inhibition of NMDARs (see Chapter 4) indicated that ethanol exerts its effect through an allosteric mechanism by reducing agonist efficacy [86].
Single channel behavior of NMDARs from hippocampal CA1 neurons was studied using very low glutamate concentrations to improve temporal resolution of individual glutamate binding events [87]. Openings resulting from individual receptor activations showed surprising complexity: they consist of a long cluster of bursts of openings. Furthermore, the NMDARs appeared to have different gating modes, occasionally entering periods of very high open probability [87]. These results demonstrated that the slow deactivation kinetics of the NMDAR result from intrinsic gating properties.
Single channel analysis can also provide more insight in how NMDARs function at the synapse. In response to a brief pulse of glutamate, mimicking synaptic release, NMDARs activate slowly over hundreds of milliseconds and continue activating long after all glutamate has been removed from the synaptic cleft, thereby briefly “memorizing” the occurrence of a synaptic input. Single channel analysis of NR1 and NR2A receptors indicates that after a brief pulse of glutamate, receptors enter a high affinity closed state from which either channel opening or agonist unbinding occurs with approximately equal probability [88]. A single synaptic event is therefore expected to only partially activate NMDARs. Consequently, a closely spaced second pulse of agonist is able to further increase the open probability, endowing the NMDAR with an ability to decode synaptic input frequency (Figure 13.1).
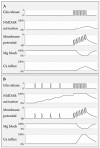
FIGURE 13.1
Decoding of synaptic inputs by NMDARs. Because of their high glutamate affinity and relatively slow activation and deactivation kinetics, NMDARs can “memorize” recent low frequency synaptic inputs that alone cannot elicit ion permeation (more...)
Comparison of the single channel behavior of NR2A- and NR2B-containing NMDARs revealed several kinetic differences in these receptor subtypes. NR2A receptors respond faster to brief synaptic-like pulses of glutamate and reach higher open probabilities [89]. It has been proposed that these differences in channel gating kinetics result in preferential opening of NR2A-containing receptors during high frequency synaptic inputs that stimulate LTP. Conversely, NR2B receptors are associated with lower frequency inputs that cause LTD. However, the NR2A/LTP and NR2B/LTD connections were discredited by recent experiments [54].
13.2.6. Subconductance Levels, Permeation, and Gating
One of the more intriguing aspects of NMDAR single channel behavior is that it is not strictly binary. Channels occasionally fail to open or close fully and instead visit intermediate conductance levels. Such subconductances levels (sublevels) have been observed in both native [90] and recombinant glutamate receptors [91–93]. Typically, these sublevels are visited during transitions between closed and fully open states, not as isolated opening events. NR2A- and NR2B-containing receptors display similar short-lived sublevels whose conductance is approximately 80% of the main open state. In contrast, NR2C- and NR2D-containing receptors briefly visit a 50% sub-level and a fully open state with approximately equal probability.
Subconductance levels have been observed in virtually every type of ion channel, although the number of levels, stability, and abundance vary widely. Sublevels have been well-characterized in K channels, which are evolutionarily related to NMDARs [94]. Our laboratory has described sublevels observed during open–closed transitions in the voltage-gated K channel Kv2.1 (drk1),95 and we have proposed a model that attributes sublevels to heteromeric pore conformations visited when one, two, or three of the four subunits move to the open conformation (Figure 13.2). Because channel opening appears to be a strongly cooperative process, the heteromeric pore conformations are predicted to be highly unstable and the associated sub-levels may be very short-lived.
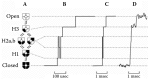
FIGURE 13.2
Subunit-subconductance model for NMDAR gating. The permeation pathway of the NMDAR is formed by two NR1 and two NR2 subunits (A). The conformation of each subunit alters between a “closed” conformation that does not support permeation (more...)
One prediction of the subunit–sublevel hypothesis is that sublevels should be more abundant when a receptor or channel is incompletely activated, which was experimentally confirmed for Kv2.1.95 In a more stringent test of this hypothesis, a Kv2.1 tandem dimer combining two K channel subunits with different activation thresholds was created. Single channel behavior of this tandem dimer at potentials between the two thresholds was dominated by two sublevels, whose kinetics and voltage dependence indicated that they resulted from the activation of one and two subunits [96]. These results support the subunit–subconductance hypothesis, which implies that gating and permeation are strictly coupled. In this model, the selectivity filter is directly responsible for opening and closing the channel [97,98]. A specific mechanism has been proposed for how the selectivity filter may function as the channel gate, in which the filter alternates between two conformations with high and low affinity for the permeant ion [98]. Experimental support for an affinity-switching selectivity filter has recently been shown by NMR experiments using the KcsA K channel [99].
Single channel analysis of AMPARs and NMDARs also provided evidence for a strong coupling between permeation and gating, as well as support for a link between sublevels and partial receptor activation. Two sublevels associated with partially activated GluR3 AMPARs were proposed to result from the activation of two and three subunits [100]. Mutations in the selectivity filter were shown to stabilize sublevels both in K channels [101,103] and NMDARs [102,104]. The ion selectivity of the sub-levels in these mutant NMDARs is different from that of the fully open state [104], as was reported for the Shaker K channel [101], thereby providing a direct causal linkage between subconductance gating and the selectivity filter.
Under bi-ionic (Na+/Cs+) conditions, gating of these mutant NMDARs becomes strongly asymmetric, with sublevels visited either during openings (external Cs+) or during closings (external Na+) [104], providing additional evidence for a strong coupling of gating and permeation. Finally, partial agonists were shown to determine the open probability of subconductance levels in the GluR2 AMPAR [105], and a model was suggested which is identical to the subunit–subconductance model (Figure 13.2) proposed by Chapman and VanDongen in 1997 [97,95]. Section 13.3.7 addresses the structural basis of partial agonism in more detail. The next section discusses the structures of NMDARs and their mechanisms of activation.
13.3. STRUCTURE AND ACTIVATION
Functional NMDARs generally form as heterotetramers of two glycine-binding NR1 subunits and two glutamate-binding NR2 subunits assembled around a central permeation pathway. The inhibitory NR3 subunit, which also binds glycine, can substitute for one NR2 subunit or replace both to form a glycine-activated receptor, although the result would not technically be considered an NMDAR. Experiments in which NMDAR subunits were covalently linked as tandem dimers suggested that the heteromeric tetramer assembles according to an NR1–NR1–NR2–NR2 arrangement [106]. Assembly of the receptor complex is thought to proceed via a “dimer-of-dimers” mechanism. Whether the initial assembly step involves homomeric or heteromeric dimers [107,108] remains controversial. Furthermore, the NR1 and NR2 subunits exhibit significant sequence homology with each other and other iGluRs, and are therefore expected to adopt similar overall domain structures.
13.3.1. Domain Structure of Subunits
Ionotropic glutamate receptor subunits are organized into four discrete functional domains (Figure 13.3): an extracellular N-terminal domain (NTD), a ligand binding domain (LBD), a pore forming transmembrane region, and an intracellular C-terminal domain. Hydrophobicity analysis performed after expression cloning of the first iGluR (AMPAR GluR1) [109] identified four hydrophobic segments (M1 through M4), the first three of which are closely spaced and separated by a long linker from the fourth segment.
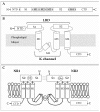
FIGURE 13.3
Domain structure of the NMDAR. A: Modular nature of linear amino acid sequences of NMDAR subunits: four hydrophobic domains (M1 through M4), two ligand binding domains (S1 and S2), and amino and carboxy terminal domains (NTD, CTD). B: Membrane topology (more...)
The three-plus-one hydrophobicity profile is very similar to previously characterized ligand gated ion channels that include the nicotinic acetylcholine receptor (nAChR) family, GABAA, glycine, and 5-HT3 receptors. It was therefore initially assumed that iGluRs would have the same membrane topology as these receptors, in which each hydrophobic domain is thought to form an α helix that crosses the membrane once [109,110]. The N-termini of all ligand-gated ion channels contain signal peptides, ensuring their extracellular localization. Therefore, in the membrane topology model for the nAChR super-family, the linker between the third and fourth transmembrane segment is predicted to be cytoplasmic.
The similarities of the nAChR and iGluR hydrophobicity profiles were later proven to be “red herrings.” A functional N-glycosylation site was identified in the M3–M4 linker of the GluR6 kainate receptor [111,112]. Since the enzymes responsible for sugar modification of asparagine residues reside in the lumen of the Golgi, N-glycosylation is a reliable marker of extracellular localization. Initially, a new topology model was proposed with an additional transmembrane segment in the middle of the M3–M4 linker [111,112]. However, functional N-glycosylation was also observed in the M3–M4 linker of kainate receptors from goldfish brain, and modification was not affected by deletion of the M2 segment, prompting the authors to conclude that M2 does not cross the membrane and the M3–M4 linker is extracellular [113].
Similar results were obtained from N-glycosylation studies of the GluR1 AMPAR and NR1 NMDAR subunits, confirming that the M2 segment does not cross the membrane [94,114]. Mutagenesis experiments on the NMDAR NR1 subunit identified residues in the M3–M4 linker critical for binding of glycine site ligands, indicating it must be extracellular [115]. An identical conclusion was reached by exchanging domains between the GluR3 AMPAR and GluR6 kainate receptor [116]. These studies also observed that two glutamate receptor domains display homology to bacterial periplasmic amino acid–binding proteins for which X-ray crystal structures already existed. This homology was first noted by Nakanishi et al. [117] and extended by O’Hara et al. [118] The first domain (S1) immediately precedes M1, while the second (S2) comprises most of the M3–M4 linker [115,116] (Figure 13.3A).
An extracellular location of the M3–M4 segment implies that the first three hydrophobic domains must span the membrane an even number of times. This discrepancy was explained by the significant amino acid sequence homology discovered between the M2 segments of iGluRs and the pore forming P regions of K channels [94], which fold as a reentrant hairpin loop. Figure 13.3 illustrates the current model for the membrane topology of NMDAR subunits [94,114].
The extracellular domains come together to form a ligand binding domain (LBD) consisting of the S1 segment preceding M1 and the S2 segment sandwiched between M3 and M4. The NTD is located N terminal to S1 and exhibits homology with metabotropic glutamate receptor (mGluR) binding domains, suggesting that it forms a ligand binding structure distinct from the agonist binding S1–S2 domain. In the NR2 subunits, the NTD has been proposed to bind zinc ions (NR2A) or polyamines (NR2B), both of which modulate NMDAR function (see Chapters 11 and 12). The ligand for the NTD of the NR1 subunit, if one exists, is currently not known.
13.3.2. Structure of the Ligand Binding Domain
The modular nature of glutamate receptors prompted experiments to isolate the ligand binding module as a separate soluble protein. Early experiments with the GluR4 AMPAR demonstrated that the isolated LBD consisting of the S1 and S2 domains connected by a short peptide linker retained the pharmacology of the full-length receptor, binding antagonists and agonists with normal affinity [119]. This approach enabled the crystallization of the GluR2 AMPAR LBD in complex with kainate [120], illustrated in Figure 13.4A.

FIGURE 13.4
(See color insert following page 212.) Crystal structures of GluR2 and NR2A ligand binding domains. X-ray crystallographic structures are shown for the isolated ligand binding domains (LBDs) of the AMPAR GluR2 in complex with kainate (A) and the NMDAR (more...)
The bilobate structure consists of two globular domains or lobes (D1 and D2), connected by a flexible hinge, and bears a striking similarity to the bacterial periplasmic amino acid–binding proteins. The S1 segment forms lobe 1 (purple) and hinge 1 (blue) that connect to the first transmembrane segment M1 in the intact receptor. S2 forms lobe 2 (green) and hinge 2 (yellow), which crosses back to lobe 1. Two helices (J and K, gray) follow hinge 2, running across the backs of the two lobes and connecting to the M4 segment. The kainate agonist is sandwiched between the two domains and forms hydrogen bonds with both lobes, thereby stabilizing the closed cleft conformation.
GluR2 structures were obtained in the absence of ligand (apo) and in complex with an antagonist (DNQX), the partial agonist kainate, and full agonists AMPA and glutamate [121]. Separation of the two domains was significantly increased in the apo state, and all ligands tested produced some amount of cleft closure. The degree of domain closure increased as follows: apo < DNQX < kainate < AMPA = glutamate. This result led to the suggestion that agonist-induced domain closure activates the channel and that degree of domain closure determines the extent of activation. Although the competitive DNQX antagonist still produces a small amount of domain closure relative to the apo state, it apparently is not sufficient to activate the channel. Section 13.3.7 discusses the structural basis for partial agonism in more detail.
Crystallographic structures for the S1–S2 LBD were obtained for additional members of the iGluR family including the GluR5 and GluR6 kainate receptors [122], GluR0 [123], a prokaryotic glutamate-gated K channel, and the NMDAR subunits NR1 [124] and NR2A [125]. All LBDs have the same basic structure, although certain conformational differences exist between the lobes and hinge regions. Figure 13.4C shows the LBD structure of the NMDAR NR2A subunit in complex with glutamate; this can be compared with the structure of GluR2 in Figure 13.4A. The availability of high-resolution crystal structures greatly facilitates the design of specific point mutations and the interpretations of resulting phenotypes.
13.3.3. Affinity and Efficacy
The interaction of a ligand with its receptor is characterized by two fundamental pharmacodynamic properties: affinity and efficacy. Affinity measures how tightly a ligand binds to a receptor and is characterized by the equilibrium dissociation constant, KD. Efficacy measures how effectively a ligand, once bound, activates the receptor. Antagonists have no efficacy. The efficacies of partial agonists are lower than those of full agonists binding to the same site. The processes of agonist binding and receptor activation are strongly coupled, with the former initiating the latter.
This coupling complicates the interpretation of phenotypes caused by mutations. A point mutation that causes a shift in the concentration–response curve can do so without affecting binding, because the midpoint of the curve (EC50) is dependent on both the binding equilibrium (affinity) and the activation process (efficacy) [126]. Examples are provided by the previously published mutations in the pore of the nicotinic acetylcholine receptor, many of which altered acetylcholine sensitivity by affecting open state stability [127,128]. Another complication is that a change in efficacy caused by a mutation does not necessarily result in a measurable change in maximum response. When efficacy is very high, even substantial changes in efficacy exert minimal effects on maximum response. Moreover, when a change in maximum response is observed for a mutation, it is often difficult to exclude altered protein folding efficiency, protein stability, and inefficient expression as possible explanations.
To solve this dilemma, we developed an approach that allows the evaluation of affinity and efficacy roles for individual amino acid positions in a receptor. First, the position to be investigated is mutated to cysteine, which ideally should have little effect on receptor pharmacology. Second, concentration–response curves are collected for a full agonist and a partial agonist before and after covalent modification of the introduced cysteine [129]. Due to their small size, MTS (methanethiosulfonate) compounds are very useful for this purpose. These experiments yield values for the EC50 and intrinsic activity (α), a measure of relative efficacy, before and after covalent modification of the cysteine.
The in situ mutagenesis produced by MTS modification does not alter the population of receptors studied because it typically takes less than a minute to complete; therefore, any change in maximum response must result from an alteration in efficacy. Using a partial agonist guarantees that any changes in efficacy caused by MTS modification will reflect a change in intrinsic activity. Based on the values of EC50 and α before and after MTS modification, it is possible to quantitate changes in affinity and efficacy caused by the modification [129].
Using this approach, Kalbaugh et al. assigned affinity and efficacy roles to positions in the LBDs of the NR1 and NR2A NMDAR subunits. In situ mutation of residues in direct contact with bound ligand affected both efficacy and affinity, while positions that stabilize the closed cleft conformation without a direct ligand interaction contributed only to efficacy [129]. These results provide a molecular basis for the tight coupling of agonist binding and receptor activation. The same residues that mediate stabilization of the closed cleft conformation by the bound ligand are also critical for ligand binding to the apo state, resulting in an agonist-bound open cleft conformation.
Crystallization of the isolated and soluble S1 and S2 LBDs for several iGluRs has significantly increased our understanding of the molecular bases of ligand binding and receptor activation mechanisms. The structure and function of the membrane domain (M1 through M4) will be discussed next.
13.3.4. Structure of the Membrane Domain: K Channel as Model
High-resolution structures are available for many LBDs of iGluRs but intact receptors, which are large integral membrane proteins, have thus far resisted crystallization. However, the M1–M3 segment appears to be structurally and functionally related to K channels, for several of which X-ray structures do exist [130–132]. Wo and Oswald (1994) originally hinted at a structural relationship between K channels and iGluRs by suggesting that the kainate receptor M2 segment folds back into the membrane similar to the P regions of K channels [113] (Figure 13.5A). N-glycosylation experiments in the M3–M4 linker by Wood et al. (1995) were prompted by a high degree of amino acid homology between NMDAR M2 segments and K channel P regions [94].

FIGURE 13.5
Relationship of glutamate receptors and K channels. A: Comparison of membrane topologies of ionotropic glutamate receptors, the prokaryotic K channel KcsA, and voltage-gated K channels. A common pore forming motif (M1–M2–M3, TM1-P–TM2, (more...)
Interestingly, the K channel GYGD signature sequence that forms part of the selectivity filter is replaced in NR1 with GIGE (Figure 13.5B). However, transplantation of the selectivity filter sequence TVGYG from K channels into iGluRs failed to transfer K selectivity to these channels, although functional channels were obtained and many pore properties were altered [133]. If the transplanted TVGYG regions fold similarly in iGluRs and K channels, as suggested by the functionality of the chimeras, it is likely that permeating ions are coordinated by backbone carbonyls in both channel families. Because iGluRs do not distinguish between Na+ and K+ ions and some members conduct divalent Ca2+ ions, the atomic distance between backbone carbonyls forming the oxygen cage should be significantly more flexible and dynamic in these nonselective cation channels.
The amino acid homology in the pore-forming regions prompted us to suggest that K channels and iGluRs have a common evolutionary ancestor [94]. This is supported by the identification of a “missing link,” the prokaryotic GluR0 glutamate receptor that has a pore-forming domain with high homology to K channel P regions and exhibits K+ selectivity [134]. Using the GluR0 amino acid sequence as a guide, the homology of K channels and iGluRs can be extended to the transmembrane domain following the pore region: M3 in iGluRs and S6 or TM2 in K channels [135] (Figure 13.5B). In K channels, the S6 and TM2 segments undergo conformational changes during channel activation [136–138]. Evidence suggests that the iGluR M3 segment may play a similarly important role during receptor activation.
13.3.5. Role of the M3 Domain
The M3 segment contains a 9-amino acid sequence (SYTANLAAF) that is highly conserved among glutamate receptors; many cysteine substitutions in this region display state-dependent accessibility [135,139]. Altered residue accessibility often indicates a conformational change [140], suggesting that the SYTANLAAF region moves in response to receptor activation; thus, M3 has been proposed to function as a transduction element, coupling ligand-binding to channel gating [135]. A recent study suggests that M3 is the only transmembrane domain contributing to the deepest portion of the pore, supporting a prominent role in gating [141].
The functional importance of the SYTANLAAF motif was originally identified in the GluRδ2 receptor, an orphan receptor with homology to the iGluR family, but no known glutamatergic agonist. A single-point mutation, A8T, was found to cause an inherited neurological defect in mice [142]. Known as lurcher, the phenotype, characterized by ataxia and neurodegeneration, is caused by constitutive activation of δ δ2 receptors, which produces excitotoxicity and apoptosis of cerebellar Purkinje cells. Introduction of the lurcher mutation in GluR1 and GluR6 produced some constitutive activity, increased agonist potency, slower deactivation, and conversion of an antagonist into an agonist [143–145]. The same substitution in NR1 and NR2A did not produce constitutive activity, but exhibited very slow deactivation; interestingly, increased glutamate potency was observed only in the NR1 mutant [143].
Similar to the lurcher phenotype, constitutive activity and/or current potentiation were observed upon thiol modification of substituted cysteines at several positions within the SYTANLAAF region [135,139,146–148]. Of particular interest is the A7C substitution in NR1 and NR2 that appears to lock the channel in a fully activated conformation upon modification [135,139,149]. A7C can be modified only after receptor activation and accessibility exhibits a linear correlation with agonist efficacy [135], strongly suggesting a role for M3 in coupling ligand binding to channel gating. Conservation of the A7C phenotype is observed between NR1 and NR2 and across NR2 subtypes, implying functional conservation of M3 among all NMDARs [139].
Several lines of evidence suggest that the NR1 and NR2 M3 domains may play distinct roles in receptor activation. NR1 and NR2 contribute differently to the M2 loop [150] that forms the inner pore of the channel, and copper coordination of substituted cysteines indicates that their M3 domains may be staggered by a full α helical turn [151]. Consistent with a dimer-of-dimers arrangement, the extracellular vestibule of the homomeric AMPAR exhibits two-fold rotational symmetry, as opposed to the four-fold symmetry of K+ channels [152]. Recent work evaluating the voltage dependence of cysteine modification suggests that the NR1 M3 domain contributes mainly to the deep portion of the pore, while the NR2 M3 comprises more of the shallow extracellular vestibule [141].
These subtle structural differences among subunits may be critical for NMDAR activation; for example, kinetic modeling with partial agonists revealed that distinct fast and slow pregating conformational changes are mediated by NR1 and NR2, respectively [153]. Residues displaying state-dependent accessibility were found to cluster together on helical net diagrams, opposite from putative pore-lining residues, suggesting that positions exhibiting lurcher-like phenotypes may be located at dynamic interfaces between transmembrane segments [141]. Thus intersubunit interactions at the transmembrane level may be critical for NMDAR activation.
13.3.6. Dimerization of Ligand Binding Domains
One of the most intriguing findings from iGluR structural studies is the tendency of S1S2 LBDs to crystallize as homomeric dimers, initially observed in a set of GluR2 crystal structures [121]. In vitro, isolated iGluR LBDs dimerize with a dissociation constant in the millimolar range, most likely due to high protein concentrations. This effect may be mimicked in vivo by association of the N terminal domains (NTDs) that occurs at lower protein concentrations [154]. In AMPARs and kainate receptors, the dimer interface is formed exclusively by hydrogen bonds, salt bridges, and hydrophobic interactions between the D1 domains, burying 900 to 1600 Å2 of solvent-accessible surface area [121,155]. The D2 domain linking the LBDs to the ion channel portion of the receptor is free to change conformation, suggesting a mechanism by which LBD conformation may be coupled to channel gating.
The functional importance of the LBD dimer interface was initially demonstrated via site-directed mutagenesis, which was used to modulate interactions at the GluR2 interface. Single-point mutations were identified which either attenuated or increased desensitization. Crystallography, ultracentrifugation, and electrophysiological studies established that dimer stability inversely correlates with extent of desensitization [156]. Subsequent studies confirmed and extended this paradigm in AMPARs and kainate receptors [157,158].
Domain–domain (D1-D1) separation associated with GluR2 desensitization has been estimated to be between 12.4 and 16.2 Å at the top of the interface, based on modification of cysteine interface mutants via bifunctional crosslinkers. Identification of a GluR2 mutant stabilized in the desensitized state (S729C) led to a crystal structure depicting a relaxed, destabilized dimer interface [158]. Crystal structures of GluR2 in complex with several positive allosteric modulators revealed that these molecules exert their potentiating effects via interface stabilization. Cyclothiazide, which blocks receptor desensitization, binds near the edge of the interface, while aniracetam stabilizes the open state by binding near the LBD hinge [156,159].
The NR1 subunit repeatedly crystallized in monomeric form until the recent cocrystallization of NR1 and NR2A revealed the existence of a heteromeric LBD dimer interface [125]. In contrast to other iGluRs, the LBD dimer interface of the NMDAR contains both D1 and D2 interactions, providing additional opportunities for intersubunit coupling. Furthermore, a critical tyrosine residue in NR1 (Y535) occupies a site homologous to the GluR2 aniracetam binding site and the sizes of substituted amino acids at this position were found to inversely correlate with deactivation rate. Thus, Y535 appears to function as an endogenous positive allosteric modulator. This key difference in interface stability has been proposed to underlie the slow deactivation of NMDARs required for their role in synaptic transmission [160], suggesting a novel modulatory site for NMDAR-based therapeutics.
The NTDs are clamshell-shaped structures, known to interact with each other and modulate the LBDs, suggesting a possible extension of the interface model [154]. Zn2+ binding to the NR2A NTD is allosterically coupled to the glutamate-binding domain. Similar results were obtained for ifenprodil in NR2B [161,162]. An NTD dimer interface may provide the stability for binding of negative modulators to effectively couple to LBDs.
13.3.7. Structural Basis of Partial Agonism
Crystal structures of the GluR2 S1S2 domain, in complex with a range of full and partial agonists, established a strong correlation between agonist efficacy and degree of LBD closure. A similar relationship was observed in the GluR6 kainate receptor [105,122]. These results led to a structural model of iGluR activation, in which agonist binding induces LBD closure by rotating D2 toward D1, separating the linker regions and promoting channel opening. According to this hypothesis, partial agonists induce less domain closure and consequently less linker separation, slowing a subunit-specific pregating conformational change [163].
Engineering steric clashes in the LBD, thus destabilizing the closed cleft state, reduces agonist efficacy and apparent affinity in NR2, GluR2, and GluR6 [164–166], while elimination of agonist–cleft clashes in GluR2 reportedly slows receptor deactivation and increases affinity and efficacy [167]. Stabilization of the NR2 closed cleft state via modulation of an endogenous D1–D2 interaction can also increase open probability, kinetically linking LBD closure and channel gating [168]. Interestingly, crystal structure data obtained from mutagenesis studies has not always followed the cleft closure–agonist efficacy correlation; GluR2 L650T, for example, stabilizes the closed cleft state, increasing kainate-induced efficacy and degree of domain closure, but the AMPA-bound structure revealed both partially and fully closed conformations [167]. A cleft stabilizing GluR6 D1–D2 interaction dramatically increased glutamate sensitivity and slowed deactivation when introduced in GluR2, but did not affect cleft closure. Full agonist-bound GluR6 is almost 6° more closed relative to GluR2 [164].
Crystal structures obtained for the NR1 subunit revealed that full and partial agonists adopt similar degrees of domain closure, indicating that partial agonist action at an NMDAR may follow a different structural paradigm from other iGluRs [169]. Because no partial agonist-bound crystal structure is presently available for the NR2 subunit, it is not yet known which model describes the behavior of the NR2 LBD. Both structural and molecular dynamics studies suggest that the LBD hinge region, particularly the second interdomain β strand, changes conformation according to agonist efficacy [169,170]. Helix F that forms part of the LBD dimer interface has been implicated in coupling agonist efficacy to channel gating in NR2A [163]. NR1-site partial agonism has also been correlated with increased inter-pocket motion, and both glycine and DCS can reportedly move within the pocket without affecting domain closure [170].
Incomplete and unstable cleft closures have been proposed to affect single channel gating similarly [166], suggesting that degree of cleft closure may simply be a physical readout of closed cleft stability. Thus, closed cleft stability, and not simply a physical change in conformation between D1 and D2, may be the principal determinant of agonist efficacy at iGluRs.
Understanding the structural basis of partial agonism is of both scientific and clinical interest, since NMDAR hypofunction has been shown to cause cognition and memory defects in animal models [171,172]. Glutamatergic dysfunction has been implicated in the pathophysiology of schizophrenia [173] and receptor augmentation may prove therapeutically valuable. Although both glycine and D-cycloserine (DCS), a well-tolerated partial glycine site agonist, initially exhibited some efficacy in the treatment of schizophrenia [174–177], a recent multisite double blind randomized trial found both glycine and DCS ineffective for treating negative symptoms and cognitive impairments [178]. DCS has been shown to enhance learning and memory performance in adult and aged rodents [179–182] and monkeys [183], and is undergoing testing as a cognitive enhancer for treating Alzheimer’s disease [184–186], head trauma [187], and fear extinction [188–193]. Reported subtype-specific partial agonism at the NMDAR also opens the possibility of individually targeting NR2 subtypes that exhibit regional and developmental variations in expression [89].
13.3.8. Positive Allosteric Modulators
Few positive modulators, endogenous or otherwise, have been found to act on the extracellular portions of NMDARs. Polyamines such as spermine and spermidine exert both inhibitory and stimulatory effects, depending on concentration and subunit composition (see Chapter 11). Polyamines are generally found within cells and whether they participate in physiological regulation of NMDARs [194] remains unclear. Pregnenolone sulfate (PS), a neurosteroid synthesized de novo in the CNS, also exhibits subunit-specific potentiation and inhibition [195,196]. A recent study suggests that PS is released in an activity-dependent manner and may attain synaptic concentrations in the micromolar range, suggesting a possible endogenous modulatory role [197].
PS potentiates NR2A- and NR2B-containing NMDARs via increases in channel open probability; however, due to a glutamate-induced reduction of receptor affinity for PS, the effect is relatively transient [198]. In contrast, NMDARs containing NR2C or NR2D subunits are inhibited by PS, and all four NR2 subtypes are inhibited by pregnanolone sulfate, a closely related neurosteroid [196]. Planar and bent ring structures appear to favor stimulation and inhibition, respectively, while both necessitate a negatively charged C3 moiety [199]. PS binding has been localized to a steroid modulatory domain, SMD1, comprised of the M4 transmembrane domain and helix J/K located in the S2 segment of the ligand binding domain [200]. Since PS is a charged molecule and potentiation displays no voltage dependence [201], helix J/K is a more likely candidate for direct binding. This region of S2 has recently been shown to form part of the LBD dimer interface, a region identified as a binding site for numerous positive allosteric modulators of AMPARs [159].
PS treatment of neuronal cultures exacerbates NMDA-induced excitotoxicity [199], while intracerebroventricular administration in mice has been shown to cause convulsions, although that effect may be due to inhibitory actions at the GABAA receptor [202]. Hippocampal PS concentration has been correlated with preserving cognitive functions in aging animals [203] and in vivo infusion of PS enhances neurogenesis in mice [204]. PS has also been shown to increase LTP in the hippocampus [205], possibly providing a mechanism for its role as a cognitive enhancer.
Although the nonselective effects of PS may preclude its clinical use, it may serve as a promising starting point for therapeutic development. Positive allosteric modulators of AMPARS, known as ampakines, improve learning and memory and suppress symptoms of schizophrenia, depression, and ADHD in animal models. Initial studies in humans have also shown positive effects on memory and psychiatric symptoms [206]. Crystal structures of GluR2 in complex with two ampakines, aniracetam and CX614, revealed that both modulators bind within the LBD dimer interface, slowing deactivation and/or desensitization [159]. Ampakines are proposed to enhance cognitive function in a three-fold manner: expansion of cortical networks, facilitation of LTP, and upregulation of BDNF expression [206]. Based on the structural homology among iGluRs, and the role of NMDARs in LTP induction, the development of an NMDAkine is certainly within the realm of possibility.
13.4. CONCLUSIONS
NMDARs play critical roles in both the proper development of the central nervous system and the processes underlying functional and structural plasticity in the adult brain. They are able to perform these tasks because they possess a combination of unique properties: (1) high affinity for the excitatory transmitter L-glutamate, (2) very slow kinetics of (de)activation, (3) pronounced voltage dependence due to external Mg block, (4) high permeability to Ca ions, and (5) large cytoplasmic domains that enable them to become part of and help organize large macromolecular synaptic signaling complexes.
The high affinity for glutamate and relatively slow (de)activation kinetics allow NMDARs to decode synaptic input patterns over prolonged periods (Figure 13.1). Their voltage dependence enables them to act like molecular coincidence detectors, mediating calcium infiux only when strong membrane depolarization coincides with synaptic release of glutamate. Ca ions entering through NMDARs act locally at signaling complexes associated with the receptor, to allow long-lasting modification of individual synapses. In addition to this highly localized action, activation of NMDARs in distal dendrites can signal to the nucleus to affect gene transcription by mechanisms that are not fully understood. Finally, the regenerative properties of NMDARs allow them to help generate and propagate dendritic depolarizations, resulting in nonlinear processing of synaptic inputs that may endow neurons with novel computational abilities.
Based on these unique properties, it is not surprising that NMDAR hypofunction or overstimulation can result in many cognitive defects and brain dysfunction, making these receptors prime therapeutic targets. Unfortunately, clinical results with NMDAR drugs have been limited because of a combination of lack of efficacy and unacceptable side effects. However, recent progress in elucidating the molecular mechanisms underlying activation of NMDARs, driven by the availability of high resolution X-ray structures for the ligand binding domains, is likely to revitalize the search for more effective and subtype-specific NMDAR drugs. Particularly promising are the allosteric modulators that either enhance or inhibit NMDAR function to a well-defined extent, without acting as (ant)agonists. Their inherent dose limiting ability indicates that allosteric modulators will be far better tolerated than the compounds investigated to date.
REFERENCES
- 1.
- Kemp JA, McKernan RM. NMDAR pathways as drug targets. Nature Neurosci. 2002;5:1039. [PubMed: 12403981]
- 2.
- Jansen M, Dannhardt G. Antagonists and agonists at the glycine site of the NMDAR for therapeutic interventions. Eur J Med Chem. 2003;38:661. [PubMed: 12932897]
- 3.
- Chazot PL. The NMDAR NR2B subunit: a valid therapeutic target for multiple CNS pathologies. Curr Med Chem. 2004;11:389. [PubMed: 14965239]
- 4.
- Farlow MR. NMDAR antagonists. A new therapeutic approach for Alzheimer’s disease. Geriatrics. 2004;59:22. [PubMed: 15224791]
- 5.
- Wood PL. The NMDAR complex: a long and winding road to therapeutics. IDrugs. 2005;8:229. [PubMed: 15772895]
- 6.
- Cai SX. Glycine/NMDAR antagonists as potential CNS therapeutic agents: ACEA-1021 and related compounds. Curr. Topics Med. Chem. 2006;6:651. [PubMed: 16719807]
- 7.
- Missale C, et al. The NMDA/D1 receptor complex as a new target in drug development. Curr Topics Med Chem. 2006;6:801. [PubMed: 16719818]
- 8.
- Brown DG, Krupp JJ. N-methyl-D-aspartate receptor (NMDA) antagonists as potential pain therapeutics. Curr Topics Med Chem. 2006;6:749. [PubMed: 16719815]
- 9.
- Dingledine R, et al. Glutamate receptor ion channels. Pharmacol Rev. 1999;51:7. [PubMed: 10049997]
- 10.
- Rothstein JD, et al. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713. [PubMed: 7917301]
- 11.
- Huang YH, Bergles DE. Glutamate transporters bring competition to the synapse. Curr Opin Neurobiol. 2004;14:346. [PubMed: 15194115]
- 12.
- Nowak L, et al. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462. [PubMed: 6320006]
- 13.
- Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg of NMDA responses in spinal cord neurones. Nature. 1984;309:261. [PubMed: 6325946]
- 14.
- Seeburg PH, et al. The NMDAR channel: molecular design of a coincidence detector. Recent Prog Hormone Res. 1995;50:19. [PubMed: 7740157]
- 15.
- Bliss TVP, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331. [PMC free article: PMC1350458] [PubMed: 4727084]
- 16.
- Collingridge GL, Bliss TV. Memories of NMDARs and LTP. Trends Neurosci. 1995;18:54. [PubMed: 7537406]
- 17.
- Feldman DE, Nicoll RA, Malenka RC. Synaptic plasticity at thalamocortical synapses in developing rat somatosensory cortex: LTP, LTD, and silent synapses. J Neurobiol. 1999;41:92. [PubMed: 10504196]
- 18.
- Johnston D. NMDA-receptor independent LTP. Neurochem Int. 1992;20:461. [PubMed: 1304864]
- 19.
- Kapur A, et al. L-Type calcium channels are required for one form of hippocampal mossy fiber LTP. J Neurophysiol. 1998;79:2181. [PMC free article: PMC2874953] [PubMed: 9535977]
- 20.
- Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc Natl Acad Sci USA. 1992;89:4363. [PMC free article: PMC49082] [PubMed: 1350090]
- 21.
- Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994;4:389. [PubMed: 7919934]
- 22.
- Bortolotto ZA, Fitzjohn SM, Collingridge GL. Roles of metabotropic glutamate receptors in LTP and LTD in the hippocampus. Curr Opin Neurobiol. 1999;9:299. [PubMed: 10395580]
- 23.
- Isaac J. Protein phosphatase 1 and LTD: synapses are the architects of depression. Neuron. 2001;32:963. [PubMed: 11754828]
- 24.
- Spruston N, et al. Activity-dependent action potential invasion and calcium infiux into hippocampal CA1 dendrites. Science. 1995;268:297. [PubMed: 7716524]
- 25.
- Stuart G, et al. Action potential initiation and backpropagation in neurons of the mammalian CNS. Trends Neurosci. 1997;20:125. [PubMed: 9061867]
- 26.
- Markram H, et al. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213. [PubMed: 8985014]
- 27.
- Debanne D, Gahwiler BH, Thompson SM. Long-term synaptic plasticity between pairs of individual CA3 pyramidal cells in rat hippocampal slice cultures. J Physiol. 1998;507:237. [PMC free article: PMC2230782] [PubMed: 9490845]
- 28.
- Debanne D, Gähwiler BH, Thompson SM. Asynchronous pre- and post-synaptic activity induces associative long-term depression in area CA1 of the rat hippocampus in vitro. Proc Natl Acad Sci USA. 1994;91:1148. [PMC free article: PMC521471] [PubMed: 7905631]
- 29.
- Abbott LF, Nelson SB. Synaptic plasticity: taming the beast. Nat Neurosci. 2000;3:1178. [PubMed: 11127835]
- 30.
- Kepecs A, et al. Spike-timing-dependent plasticity: common themes and divergent vistas. Biol Cybernet. 2002;87:446. [PubMed: 12461634]
- 31.
- Lisman J, Spruston N. Postsynaptic depolarization requirements for LTP and LTD: a critique of spike timing-dependent plasticity. Nat Neurosci. 2005;8:839. [PubMed: 16136666]
- 32.
- Dan Y, Poo MM. Spike timing-dependent plasticity: from synapse to perception. Physiol Rev. 2006;86:1033. [PubMed: 16816145]
- 33.
- Drew PJ, Abbott LF. Extending the effects of spike-timing-dependent plasticity to behavioral timescales. Proc Natl Acad Sci USA. 2006;103:8876. [PMC free article: PMC1470971] [PubMed: 16731625]
- 34.
- Lin YW, et al. Spike-timing-dependent plasticity at resting and conditioned lateral perforant path synapses on granule cells in the dentate gyrus: different roles of N-methyl-D-aspartate and group I metabotropic glutamate receptors. Eur J Neurosci. 2006;23:2362. [PubMed: 16706844]
- 35.
- Gerkin RC, et al. Modular competition driven by NMDAR subtypes in spike-timing-dependent plasticity. J Neurophysiol. 2007;97:2851. [PubMed: 17267756]
- 36.
- Bi G, et al. Synaptic modification by correlated activity: Hebb’s postulate revisited. Annu Rev Neurosci. 2001;24:139. [PubMed: 11283308]
- 37.
- van Rossum MC, Bi GQ, Turrigiano GG. Stable Hebbian learning from spike timing-dependent plasticity. J Neurosci. 2000;20:8812. [PMC free article: PMC6773092] [PubMed: 11102489]
- 38.
- Schiller J, et al. NMDA spikes in basal dendrites of cortical pyramidal neurons. Nature. 2000;404:285. [PubMed: 10749211]
- 39.
- Schiller J, Schiller Y. NMDAR-mediated dendritic spikes and coincident signal amplification. Curr Opin Neurobiol. 2001;11:343. [PubMed: 11399433]
- 40.
- Enoki R, et al. NMDAR-mediated depolarizing after-potentials in the basal dendrites of CA1 pyramidal neurons. Neurosci Res. 2004;48:325. [PubMed: 15154678]
- 41.
- Gordon U, Polsky A, Schiller J. Plasticity compartments in basal dendrites of neocortical pyramidal neurons. J Neurosci. 2006;26:12717. [PMC free article: PMC6674852] [PubMed: 17151275]
- 42.
- Nevian T, et al. Properties of basal dendrites of layer 5 pyramidal neurons: a direct patch-clamp recording study. Nat Neurosci. 2007;10:206. [PubMed: 17206140]
- 43.
- London M, Hausser M. Dendritic computation. Annu Rev Neurosci. 2005;28:503. [PubMed: 16033324]
- 44.
- Rhodes P. The properties and implications of NMDA spikes in neocortical pyramidal cells. J Neurosci. 2006;26:6704. [PMC free article: PMC6673826] [PubMed: 16793878]
- 45.
- Gasparini S, Magee JC. State-dependent dendritic computation in hippocam-pal CA1 pyramidal neurons. J Neurosci. 2006;26:2088. [PMC free article: PMC6674927] [PubMed: 16481442]
- 46.
- Lisman J. A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc Natl Acad Sci USA. 1989;86:9574. [PMC free article: PMC298540] [PubMed: 2556718]
- 47.
- Brickley SG, et al. NR2B and NR2D subunits coassemble in cerebellar Golgi cells to form a distinct NMDAR subtype restricted to extrasynaptic sites. J Neurosci. 2003;23:4958. [PMC free article: PMC6741215] [PubMed: 12832518]
- 48.
- Thomas CG, Miller AJ, Westbrook GL. Synaptic and extrasynaptic NMDAR NR2 subunits in cultured hippocampal neurons. J Neurophysiol. 2006;95:1727. [PubMed: 16319212]
- 49.
- Hardingham GE, Bading H. The Yin and Yang of NMDAR signalling. Trends Neurosci. 2003;26:81. [PubMed: 12536131]
- 50.
- Vanhoutte P, Bading H. Opposing roles of synaptic and extrasynaptic NMDARs in neuronal calcium signalling and BDNF gene regulation. Curr Opin Neurobiol. 2003;13:366. [PubMed: 12850222]
- 51.
- Liu L, et al. Role of NMDAR subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021. [PubMed: 15143284]
- 52.
- Massey PV, et al. Differential roles of NR2A and NR2B-containing NMDARs in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821. [PMC free article: PMC6729941] [PubMed: 15356193]
- 53.
- Berberich S, et al. Lack of NMDAR subtype selectivity for hippocampal long-term potentiation. J Neurosci. 2005;25:6907. [PMC free article: PMC6725356] [PubMed: 16033900]
- 54.
- Morishita W, et al. Activation of NR2B-containing NMDARs is not required for NMDAR-dependent long-term depression. Neuropharmacology. 2007;52:71. [PubMed: 16899258]
- 55.
- Husi H, Grant SG. Proteomics of the nervous system. Trends Neurosci. 2001;24:259. [PubMed: 11311377]
- 56.
- Husi H, et al. Proteomic analysis of NMDAR-adhesion protein signaling complexes. Nat Neurosci. 2000;3:661. [PubMed: 10862698]
- 57.
- Kennedy MB. Signal-processing machines at the postsynaptic density. Science. 2000;290:750. [PubMed: 11052931]
- 58.
- Brambilla R, et al. A role for the Ras signalling pathway in synaptic transmission and long-term memory. Nature. 1997;390:281. [PubMed: 9384379]
- 59.
- Gnegy ME. Ca2+/calmodulin signaling in NMDA-induced synaptic plasticity. Crit Rev Neurobiol. 2000;14:91. [PubMed: 11513244]
- 60.
- Sheng M. The postsynaptic NMDA-receptor-PSD-95 signaling complex in excitatory synapses of the brain. J Cell Sci. 2001;114:1251. [PubMed: 11256991]
- 61.
- Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem. 2001;76:1. [PubMed: 11145972]
- 62.
- Riccio A, Ginty DD. What a privilege to reside at the synapse: NMDAR signaling to CREB. Nat Neurosci. 2002;5:389. [PubMed: 11976696]
- 63.
- Deisseroth K, et al. Signaling from synapse to nucleus: the logic behind the mechanisms. Curr Opin Neurobiol. 2003;13:354. [PubMed: 12850221]
- 64.
- Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325:529. [PubMed: 2433595]
- 65.
- Kleckner NW, Dingledine R. Requirement for glycine in activation of NMDARs expressed in Xenopus oocytes. Science. 1988;241:835. [PubMed: 2841759]
- 66.
- Dingledine R, Kleckner NW, McBain CJ. The glycine coagonist site of the NMDAR. Adv Exp Med Biol. 1990;268:17. [PubMed: 2150150]
- 67.
- Panatier A, et al. Glia-derived D-serine controls NMDAR activity and synaptic memory. Cell. 2006;125:775. [PubMed: 16713567]
- 68.
- Ascher P. Measuring and controlling the extracellular glycine concentration at the NMDAR level. Adv Exp Med Biol. 1990;268:13. [PubMed: 2150149]
- 69.
- Hood WF, Compton RP, Monahan JB. D-cycloserine: a ligand for the N-methyl-D-aspartate coupled glycine receptor has partial agonist characteristics. Neurosci Lett. 1989;98:91. [PubMed: 2540460]
- 70.
- Henderson G, Johnson JW, Ascher P. Competitive antagonists and partial agonists at the glycine modulatory site of the mouse N-methyl-D-aspartate receptor. J Physiol. 1990;430:189. [PMC free article: PMC1181734] [PubMed: 1707965]
- 71.
- Baran H, Loscher W, Mevissen M. The glycine/NMDAR partial agonist D-cycloserine blocks kainate-induced seizures in rats: comparison with MK-801 and diazepam. Brain Res. 1994;652:195. [PubMed: 7953730]
- 72.
- Rundfeldt C, Wla P, Loscher W. Anticonvulsant activity of antagonists and partial agonists for the NMDAR-associated glycine site in the kindling model of epilepsy. Brain Res. 1994;653:125. [PubMed: 7982045]
- 73.
- Seguin L, Millan MJ. The glycine B receptor partial agonist, (+)-HA966, enhances induction of antinociception by RP 67580 and CP-99,994. Eur. J. Pharmacol. 1994;253:R1. [PubMed: 8013534]
- 74.
- Tricklebank MD, et al. The anticonvulsant and behavioural profile of L-687,414, a partial agonist acting at the glycine modulatory site on the N-methyl-D-aspartate (NMDA) receptor complex. Brit J Pharmacol. 1994;113:729. [PMC free article: PMC1510413] [PubMed: 7858861]
- 75.
- Pussinen R, et al. Enhancement of intermediate-term memory by an alpha-1 agonist or a partial agonist at the glycine site of the NMDAR. Neurobiol Learn Mem. 1997;67:69. [PubMed: 9013503]
- 76.
- Priestley T, et al. L-687,414, a low efficacy NMDAR glycine site partial agonist in vitro, does not prevent hippocampal LTP in vivo at plasma levels known to be neuroprotective. Brit J Pharmacol. 1998;124:1767. [PMC free article: PMC1565569] [PubMed: 9756395]
- 77.
- Jones KS, VanDongen HM, VanDongen AM. The NMDAR M3 segment is a conserved transduction element coupling ligand binding to channel opening. J Neurosci. 2002;22:2044. [PMC free article: PMC6758261] [PubMed: 11896144]
- 78.
- Ballard TM, et al. Severe impairment of NMDAR function in mice carrying targeted point mutations in the glycine binding site results in drug-resistant nonhabituating hyperactivity. J Neurosci. 2002;22:6713. [PMC free article: PMC6758156] [PubMed: 12151550]
- 79.
- Zylberman I, Javitt DC, Zukin SR. Pharmacological augmentation of NMDAR function for treatment of schizophrenia. Ann NY Acad Sci. 1995;757:487. [PubMed: 7611706]
- 80.
- Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDAR hypofunction to the dopamine hypothesis of schizophrenia. Neuropsycho-pharmacology. 1999;20:201. [PubMed: 10063482]
- 81.
- Coyle JT, Tsai G. NMDAR function, neuroplasticity, and the pathophysiology of schizophrenia. Int Rev Neurobiol. 2004;59:491. [PubMed: 15006500]
- 82.
- Pilowsky LS, et al. First in vivo evidence of an NMDAR deficit in medication-free schizophrenic patients. Mol Psych. 2006;11:118. [PubMed: 16189506]
- 83.
- Neher E, Sakmann B. Single-channel currents recorded from membrane of denervated frog muscle fibres. Nature. 1976;260:799. [PubMed: 1083489]
- 84.
- Wright JM, Peoples RW. NMDAR pharmacology and analysis of patch-clamp recordings. Meth Mol Biol. 1999;128:143. [PubMed: 10320980]
- 85.
- Rock DM, Macdonald RL. The polyamine spermine has multiple actions on N-methyl-D-aspartate receptor single-channel currents in cultured cortical neurons. Mol Pharmacol. 1992;41:83. [PubMed: 1370709]
- 86.
- Wright JM, Peoples RW, Weight FF. Single-channel and whole-cell analysis of ethanol inhibition of NMDA-activated currents in cultured mouse cortical and hippocampal neurons. Brain Res. 1996;738:249. [PubMed: 8955520]
- 87.
- Gibb AJ, Colquhoun D. Glutamate activation of a single NMDAR-channel produces a cluster of channel openings. Proc. R. Soc. Lond. (Biol.). 1991;243:39. [PubMed: 1708142]
- 88.
- Popescu G, et al. Reaction mechanism determines NMDAR response to repetitive stimulation. Nature. 2004;430:790. [PubMed: 15306812]
- 89.
- Erreger K, et al. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol. 2005;563:345. [PMC free article: PMC1665591] [PubMed: 15649985]
- 90.
- Jahr CE, Stevens CF. Glutamate activates multiple single channel conductances in hippocampal neurons. Nature. 1987;325:522. [PubMed: 2433593]
- 91.
- Stern P, et al. Single-channel conductances of NMDARs expressed from cloned cDNAs: Comparison with native receptors. Proc. R. Soc. Lond. (Biol.). 1992;250:271. [PubMed: 1283639]
- 92.
- Stern P, et al. Single channel properties of cloned NMDARs in a human cell line—comparison with results from Xenopus oocytes. J Physiol. 1994;476:391. [PMC free article: PMC1160453] [PubMed: 8057248]
- 93.
- Wyllie DJ, et al. Single-channel currents from recombinant NMDA NR1a/NR2D receptors expressed in Xenopus oocytes. Proc. R. Soc. Lond. (Biol.). 1996;263:1079. [PubMed: 8805841]
- 94.
- Wood MW, VanDongen HMA, VanDongen AMJ. Structural conservation of ion conduction pathways in K channels and glutamate receptors. Proc Natl Acad Sci USA. 1995;92:4882. [PMC free article: PMC41811] [PubMed: 7761417]
- 95.
- Chapman ML, VanDongen HMA, VanDongen AMJ. Activation-dependent subconductance levels in K channels suggest a subunit basis for ion permeation and gating. Biophys J. 1997;72:708. [PMC free article: PMC1185596] [PubMed: 9017198]
- 96.
- Chapman ML, VanDongen AM. K channel subconductance levels result from heteromeric pore conformations. J Gen Physiol. 2005;126:87. [PMC free article: PMC2266570] [PubMed: 16043772]
- 97.
- VanDongen AMJ. Structure and function of ion channels: a hole in four? Comm Theor Biol. 1992;2:429.
- 98.
- VanDongen AM. K channel gating by an affinity-switching selectivity filter. Proc Natl Acad Sci USA. 2004;101:3248. [PMC free article: PMC365775] [PubMed: 14976245]
- 99.
- Baker KA, et al. Conformational dynamics of the KcsA potassium channel governs gating properties. Nat Struc Mol Biol. 2007;14:1089. [PMC free article: PMC3525321] [PubMed: 17922011]
- 100.
- Rosenmund C, Stern-Bach Y, Stevens CF. The tetrameric structure of a glutamate receptor channel. Science. 1998;280:1596. [PubMed: 9616121]
- 101.
- Zheng J, Sigworth FJ. Selectivity changes during activation of mutant Shaker potassium channel. J Gen Physiol. 1997;110:101. [PMC free article: PMC2233792] [PubMed: 9236204]
- 102.
- Premkumar LS, Qin F, Auerbach A. Subconductance states of a mutant NMDAR channel kinetics, calcium, and voltage dependence. J Gen Physiol. 1997;109:181. [PMC free article: PMC2220056] [PubMed: 9041447]
- 103.
- Zheng J, Sigworth FJ. Intermediate conductances during deactivation of heteromultimeric Shaker potassium channels. J Gen Physiol. 1998;112:457. [PMC free article: PMC2229424] [PubMed: 9758864]
- 104.
- Schneggenburger R, Ascher P. Coupling of permeation and gating in an NMDA-channel pore mutant. Neuron. 1997;18:167. [PubMed: 9010214]
- 105.
- Jin R, et al. Structural basis for partial agonist action at ionotropic glutamate receptors. Nat Neurosci. 2003;6:803. [PubMed: 12872125]
- 106.
- Schorge S, Colquhoun D. Studies of NMDAR function and stoichiometry with truncated and tandem subunits. J Neurosci. 2003;23:1151. [PMC free article: PMC6742241] [PubMed: 12598603]
- 107.
- Papadakis M, Hawkins LM, Stephenson FA. Appropriate NR1-NR1 disulfide-linked homodimer formation is requisite for efficient expression of functional, cell surface N-methyl-D-aspartate NR1/NR2 receptors. J Biol Chem. 2004;279:14703. [PubMed: 14732708]
- 108.
- Schuler T, et al. Formation of NR1/NR2 and NR1/NR3 heterodimers constitutes initial step in N-methyl-D-Aspartate receptor assembly. J. Biol. Chem. 2008;283:37. [PubMed: 17959602]
- 109.
- Hollmann M, et al. Cloning by functional expression of a member of the glutamate receptor family. Nature. 1989;342:643. [PubMed: 2480522]
- 110.
- Keinanen K, et al. A family of AMPA-selective glutamate receptors. Science. 1990;249:556. [PubMed: 2166337]
- 111.
- Roche KW, et al. Transmembrane topology of the glutamate receptor subunit GluR6. J Biol Chem. 1994;269:11679. [PubMed: 8163463]
- 112.
- Taverna FA, et al. A transmembrane model for an ionotropic glutamate receptor predicted on the basis of the location of asparagine-linked oligosaccharides. J Biol Chem. 1994;269:14159. [PubMed: 8188697]
- 113.
- Wo ZG, Oswald RE. Transmembrane topology of two kainate receptor subunits revealed by N-glycosylation. Proc Natl Acad Sci USA. 1994;91:7154. [PMC free article: PMC44357] [PubMed: 8041762]
- 114.
- Hollmann M, Maron C, Heinemann SF. N-glycosylation site tagging suggests a three transmembrane domain topology for the glutamate receptor GluR1. Neuron. 1994;13:1331. [PubMed: 7993626]
- 115.
- Kuryatov A, et al. Mutational analysis of the glycine-binding site of the NMDAR: structural similarity with bacterial amino acid-binding proteins. Neuron. 1994;12:1291. [PubMed: 8011339]
- 116.
- Stern-Bach Y, et al. Agonist selectivity of glutamate receptors is specified by two domains structurally related to bacterial amino acid–binding proteins. Neuron. 1994;13:1345. [PubMed: 7527641]
- 117.
- Nakanishi N, Shneider NA, Axel R. A family of glutamate receptor genes: evidence for the formation of heteromultimeric receptors with distinct channel properties. Neuron. 1990;5:569. [PMC free article: PMC4481242] [PubMed: 1699567]
- 118.
- O’Hara PJ, et al. The ligand binding domain in metabotropic glutamate receptors is related to bacterial periplasmic binding proteins. Neuron. 1993;11:41. [PubMed: 8338667]
- 119.
- Kuusinen A, Arvola M, Keinanen K. Molecular dissection of the agonist binding site of an AMPA receptor. EMBO J. 1995;14:6327. [PMC free article: PMC394757] [PubMed: 8557052]
- 120.
- Armstrong N, et al. Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature. 1998;395:913. [PubMed: 9804426]
- 121.
- Armstrong N, Gouaux E. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron. 2000;26:165. [PubMed: 11086992]
- 122.
- Mayer ML. Crystal structures of the GluR5 and GluR6 ligand binding cores: molecular mechanisms underlying kainate receptor selectivity. Neuron. 2005;45:539. [PubMed: 15721240]
- 123.
- Mayer ML, Olson R, Gouaux E. Mechanisms for ligand binding to GluR0 ion channels: crystal structures of the glutamate and serine complexes and a closed apo state. J Mol Biol. 2001;311:815. [PubMed: 11518533]
- 124.
- Furukawa H, Gouaux E. Mechanisms of activation, inhibition and specificity: crystal structures of the NMDAR NR1 ligand-binding core. EMBO J. 2003;22:2873. [PMC free article: PMC162155] [PubMed: 12805203]
- 125.
- Furukawa H, et al. Subunit arrangement and function in NMDARs. Nature. 2005;438:185. [PubMed: 16281028]
- 126.
- Colquhoun D. Binding, gating, affinity and efficacy: The interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Brit J Pharmacol. 1998;125:924. [PMC free article: PMC1565672] [PubMed: 9846630]
- 127.
- Akabas MH, et al. Identification of acetylcholine receptor channel-lining residues in the entire M2 segment of the alpha subunit. Neuron. 1994;13:919. [PubMed: 7524560]
- 128.
- Akabas MH, et al. Acetylcholine receptor channel structure probed in cysteine- substitution mutants. Science. 1992;258:307. [PubMed: 1384130]
- 129.
- Kalbaugh TL, VanDongen HMA, VanDongen AMJ. Ligand-binding residues integrate affinity and efficacy in the NMDAR. Mol Pharmacol. 2004;66:209. [PubMed: 15266011]
- 130.
- Doyle DA, et al. The structure of the potassium channel: molecular basis of K conduction and selectivity. Science. 1998;280:69. [PubMed: 9525859]
- 131.
- Jiang Y, et al. X-ray structure of a voltage-dependent K channel. Nature. 2003;423:33. [PubMed: 12721618]
- 132.
- Jiang Y, et al. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 2002;417:515. [PubMed: 12037559]
- 133.
- Hoffmann J, Gorodetskaia A, Hollmann M. Ion pore properties of ionotropic glutamate receptors are modulated by a transplanted potassium channel selectivity filter. Mol Cell Neurosci. 2006;33:335. [PubMed: 17010644]
- 134.
- Chen GQ, et al. Functional characterization of a potassium-selective prokaryotic glutamate receptor. Nature. 1999;402:817. [PubMed: 10617203]
- 135.
- Jones KS, VanDongen HMA, VanDongen AMJ. The NMDAR M3 segment is a conserved transduction element coupling ligand binding to channel opening. J Neurosci. 2002;22:2044. [PMC free article: PMC6758261] [PubMed: 11896144]
- 136.
- Cuello LG, et al. pH-dependent gating in the Streptomyces lividans K+ channel. Biochemistry. 1998;37:3229. [PubMed: 9536962]
- 137.
- Yellen G. The moving parts of voltage-gated ion channels. Q Rev Biophys. 1998;31:239. [PubMed: 10384687]
- 138.
- Perozo E, Cortes DM, Cuello LG. Structural rearrangements underlying K+-channel activation gating. Science. 1999;285:73. [PubMed: 10390363]
- 139.
- Yuan H, et al. Conserved structural and functional control of N-methyl-D-aspartate receptor gating by transmembrane domain M3. J Biol Chem. 2005;280:29708. [PubMed: 15970596]
- 140.
- Karlin A, Akabas MH. Substituted-cysteine accessibility method. Meth Enz. 1998;293:123. [PubMed: 9711606]
- 141.
- Sobolevsky AI, et al. Subunit-specific contribution of pore-forming domains to NMDAR channel structure and gating. J Gen Physiol. 2007;129:509. [PMC free article: PMC2151626] [PubMed: 17504910]
- 142.
- Zhou J, et al. Neurodegeneration in Lurcher mice caused by mutation in delta2 glutamate receptor. Nature. 1997;388:769. [PubMed: 9285588]
- 143.
- Kohda K, Wang Y, Yuzaki M. Mutation of a glutamate receptor motif reveals its role in gating and delta2 receptor channel properties. Nat Neurosci. 2000;3:315. [PubMed: 10725919]
- 144.
- Taverna FA, et al. The lurcher mutation of an alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor subunit enhances potency of glutamate and converts an antagonist into an agonist. J Biol Chem. 2000;275:8475. [PubMed: 10722683]
- 145.
- Klein RM, Howe JR. Effects of the lurcher mutation on GluR1 desensitization and activation kinetics. J Neurosci. 2004;24:4941. [PMC free article: PMC6729373] [PubMed: 15163686]
- 146.
- Beck C, et al. NMDAR channel segments forming the extracellular vestibule inferred from the accesibility of substituted cysteines. Neuron. 1999;22:559. [PubMed: 10197535]
- 147.
- Sobolevsky AI, Beck C, Wollmuth LP. Molecular rearrangements of the extracellular vestibule in NMDAR channels during gating. Neuron. 2002;33:75. [PubMed: 11779481]
- 148.
- Sobolevsky AI, Yelshansky MV, Wollmuth LP. Different gating mechanisms in glutamate receptor and K+ channels. J Neurosci. 2003;23:7559. [PMC free article: PMC6740752] [PubMed: 12930794]
- 149.
- Chen PE, et al. Structural features of the glutamate binding site in recombinant NR1/NR2A N-methyl-D-aspartate receptors determined by site-directed mutagenesis and molecular modeling. Mol Pharmacol. 2005;67:1470. [PubMed: 15703381]
- 150.
- Kuner T, Schoepfer R. Multiple structural elements determine subunit specificity of Mg2+ block in NMDAR channels. J Neurosci. 1996;16:3549. [PMC free article: PMC6578835] [PubMed: 8642401]
- 151.
- Sobolevsky AI, Rooney L, Wollmuth LP. Staggering of subunits in NMDAR channels. Biophys J. 2002;83:3304. [PMC free article: PMC1302406] [PubMed: 12496098]
- 152.
- Sobolevsky AI, Yelshansky MV, Wollmuth LP. The outer pore of the glutamate receptor channel has two-fold rotational symmetry. Neuron. 2004;41:367. [PubMed: 14766176]
- 153.
- Banke TG, Traynelis SF. Activation of NR1/NR2B NMDARs. Nat Neurosci. 2003;6:144. [PubMed: 12524545]
- 154.
- Mayer ML. Glutamate receptors at atomic resolution. Nature. 2006;440:456. [PubMed: 16554805]
- 155.
- Nanao MH, et al. Structure of the kainate receptor subunit GluR6 agonist-binding domain complexed with domoic acid. Proc Natl Acad Sci USA. 2005;102:1708. [PMC free article: PMC547884] [PubMed: 15677325]
- 156.
- Sun Y, et al. Mechanism of glutamate receptor desensitization. Nature. 2002;417:245. [PubMed: 12015593]
- 157.
- Zhang Y, et al. Interface interactions modulating desensitization of the kainate-selective ionotropic glutamate receptor subunit GluR6. J Neurosci. 2006;26:10033. [PMC free article: PMC6674465] [PubMed: 17005866]
- 158.
- Armstrong N, et al. Measurement of conformational changes accompanying desensitization in an ionotropic glutamate receptor. Cell. 2006;127:85. [PubMed: 17018279]
- 159.
- Jin R, et al. Mechanism of positive allosteric modulators acting on AMPA receptors. J Neurosci. 2005;25:9027. [PMC free article: PMC6725607] [PubMed: 16192394]
- 160.
- Lester RA, et al. Channel kinetics determine the time course of NMDAR-mediated synaptic currents. Nature. 1990;346:565. [PubMed: 1974037]
- 161.
- Zheng F, et al. Allosteric interaction between the amino terminal domain and the ligand binding domain of NR2A. Nat Neurosci. 2001;4:894. [PubMed: 11528420]
- 162.
- Perin-Dureau F, et al. Mapping the binding site of the neuroprotectant ifenprodil on NMDARs. J Neurosci. 2002;22:5955. [PMC free article: PMC6757916] [PubMed: 12122058]
- 163.
- Erreger K, et al. Mechanism of partial agonism at NMDARs for a conformationally restricted glutamate analog. J Neurosci. 2005;25:7858. [PMC free article: PMC6725262] [PubMed: 16120788]
- 164.
- Weston MC, et al. Interdomain interactions in AMPA and kainate receptors regulate affinity for glutamate. J Neurosci. 2006;26:7650. [PMC free article: PMC6674271] [PubMed: 16855092]
- 165.
- Hansen KB, et al. Tweaking agonist efficacy at N-methyl-D-aspartate receptors by site-directed mutagenesis. Mol Pharmacol. 2005;68:1510. [PubMed: 16131614]
- 166.
- Robert A, et al. AMPA receptor binding cleft mutations that alter affinity, efficacy, and recovery from desensitization. J Neurosci. 2005;25:3752. [PMC free article: PMC6724928] [PubMed: 15829627]
- 167.
- Armstrong N, Mayer M, Gouaux E. Tuning activation of the AMPA-sensitive GluR2 ion channel by genetic adjustment of agonist-induced conformational changes. Proc Natl Acad Sci USA. 2003;100:5736. [PMC free article: PMC156270] [PubMed: 12730367]
- 168.
- Maier W, et al. Disruption of interdomain interactions in the glutamate binding pocket affects differentially agonist affinity and efficacy of N-methyl-D-aspartate receptor activation. J Biol Chem. 2007;282:1863. [PubMed: 17105731]
- 169.
- Inanobe A, Furukawa H, Gouaux E. Mechanism of partial agonist action at the NR1 subunit of NMDARs. Neuron. 2005;47:71. [PubMed: 15996549]
- 170.
- Kaye SL, Sansom MS, Biggin PC. Molecular dynamics simulations of the ligand-binding domain of an N-methyl-D-aspartate receptor. J Biol Chem. 2006;281:12736. [PubMed: 16513640]
- 171.
- Morris RG, et al. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature. 1986;319:774. [PubMed: 2869411]
- 172.
- McHugh TJ, et al. Impaired hippocampal representation of space in CA1-specific NMDAR1 knockout mice. Cell. 1996;87:1339. [PubMed: 8980239]
- 173.
- Mouri A, et al. Phencyclidine animal models of schizophrenia: approaches from abnormality of glutamatergic neurotransmission and neurodevelopment. Neuro-chem. Int. 2007;51:173. [PubMed: 17669558]
- 174.
- Heresco-Levy U, Silipo G, Javitt DC. Glycinergic augmentation of NMDAR-mediated neurotransmission in the treatment of schizophrenia. Psychopharmacol Bull. 1996;32:731. [PubMed: 8993096]
- 175.
- Coyle JT, Tsai G. The NMDAR glycine modulatory site: a therapeutic target for improving cognition and reducing negative symptoms in schizophrenia. Psycho-pharmacology. 2004;174:32. [PubMed: 15205876]
- 176.
- Duncan EJ, et al. Effects of D-cycloserine on negative symptoms in schizophrenia. Schizophren Res. 2004;71:239. [PubMed: 15474895]
- 177.
- Heresco-Levy U, Javitt DC. Comparative effects of glycine and D-cycloserine on persistent negative symptoms in schizophrenia: a retrospective analysis. Schizophren Res. 2004;66:89. [PubMed: 15061240]
- 178.
- Buchanan RW, et al. Cognitive and Negative Symptoms in Schizophrenia Trial (CONSIST): the efficacy of glutamatergic agents for negative symptoms and cognitive impairments. Am J Psych. 2007;164:1593. [PubMed: 17898352]
- 179.
- Monahan JB, et al. D-cycloserine, a positive modulator of the N-methyl-D-aspartate receptor, enhances performance of learning tasks in rats. Pharmacol Biochem Behav. 1989;34:649. [PubMed: 2560209]
- 180.
- Billard JM, Rouaud E. Deficit of NMDAR activation in CA1 hippocampal area of aged rats is rescued by D-cycloserine. Eur J Neurosci. 2007;25:2260. [PubMed: 17445224]
- 181.
- Gabriele A, Packard MG. D-Cycloserine enhances memory consolidation of hippocampus-dependent latent extinction. Learn Mem. 2007;14:468. [PubMed: 17600116]
- 182.
- Zlomuzica A, et al. NMDAR modulation by D-cycloserine promotes episodic-like memory in mice. Psychopharmacology. 2007;193:503. [PubMed: 17497136]
- 183.
- Matsuoka N, Aigner TG. D-cycloserine, a partial agonist at the glycine site coupled to N-methyl-D-aspartate receptors, improves visual recognition memory in rhesus monkeys. J Pharmacol Exp Ther. 1996;278:891. [PubMed: 8768744]
- 184.
- Randolph C, et al. D-cycloserine treatment of Alzheimer disease. Alzheimer’s Dis Assoc Dis. 1994;8:198. [PubMed: 7986489]
- 185.
- Schwartz BL, et al. d-Cycloserine enhances implicit memory in Alzheimer patients. Neurology. 1996;46:420. [PubMed: 8614505]
- 186.
- Tsai GE, et al. Improved cognition in Alzheimer’s disease with short-term D-cyclo-serine treatment. Am J Psych. 1999;156:467. [PubMed: 10080566]
- 187.
- Yaka R, et al. D-cycloserine improves functional recovery and reinstates long-term potentiation (LTP) in a mouse model of closed head injury. FASEB J. 2007;21:2033. [PubMed: 17351125]
- 188.
- Bertotto ME, et al. Infiuence of ethanol withdrawal on fear memory: Effect of D-cycloserine. Neurosci. 2006;142:979. [PubMed: 16934411]
- 189.
- Guastella AJ, et al. A randomized controlled trial of the effect of d-cycloserine on exposure therapy for spider fear. J Psych Res. 2007;41:466. [PubMed: 16828803]
- 190.
- Ledgerwood L, Richardson R, Cranney J. D-cycloserine facilitates extinction of learned fear: effects on reacquisition and generalized extinction. Biol Psych. 2005;57:841. [PubMed: 15820704]
- 191.
- Ressler KJ, et al. Cognitive enhancers as adjuncts to psychotherapy: use of D-cyclo-serine in phobic individuals to facilitate extinction of fear. Arch Gen Psych. 2004;61:1136. [PubMed: 15520361]
- 192.
- Richardson R, Ledgerwood L, Cranney J. Facilitation of fear extinction by D-cycloserine: theoretical and clinical implications. Learn Mem. 2004;11:510. [PubMed: 15466301]
- 193.
- Weber M, Hart J, Richardson R. Effects of D-cycloserine on extinction of learned fear to an olfactory cue. Neurobiol Learn Mem. 2007;87:476. [PubMed: 17275356]
- 194.
- Munir M, Subramaniam S, McGonigle P. Polyamines modulate the neuro-toxic effects of NMDA in vivo. Brain Res. 1993;616:163. [PubMed: 8358608]
- 195.
- Wu FS, Gibbs TT, Farb DH. Pregnenolone sulfate: a positive allosteric modulator at the N-methyl-D-aspartate receptor. Mol Pharmacol. 1991;40:333. [PubMed: 1654510]
- 196.
- Malayev A, Gibbs TT, Farb DH. Inhibition of the NMDA response by pregnenolone sulphate reveals subtype selective modulation of NMDARs by sulphated steroids. Br J Pharmacol. 2002;135:901. [PMC free article: PMC1573207] [PubMed: 11861317]
- 197.
- Mameli M, et al. Neurosteroid-induced plasticity of immature synapses via retrograde modulation of presynaptic NMDARs. J Neurosci. 2005;25:2285. [PMC free article: PMC6726098] [PubMed: 15745954]
- 198.
- Horak M, et al. Molecular mechanism of pregnenolone sulfate action at NR1/NR2B receptors. J Neurosci. 2004;24:10318. [PMC free article: PMC6730288] [PubMed: 15548645]
- 199.
- Weaver CE, et al. Geometry and charge determine pharmacological effects of steroids on N-methyl-D-aspartate receptor-induced Ca(2+) accumulation and cell death. J Pharmacol Exp Ther. 2000;293:747. [PubMed: 10869372]
- 200.
- Jang MK, et al. A steroid modulatory domain on NR2B controls N-methyl-D-aspartate receptor proton sensitivity. Proc Natl Acad Sci USA. 2004;101:8198. [PMC free article: PMC419580] [PubMed: 15150412]
- 201.
- Park-Chung M, et al. Distinct sites for inverse modulation of N-methyl-D-aspartate receptors by sulfated steroids. Mol Pharmacol. 1997;52:1113. [PubMed: 9396781]
- 202.
- Kokate TG, et al. Convulsant actions of the neurosteroid pregnenolone sulfate in mice. Brain Res. 1999;831:119. [PubMed: 10411990]
- 203.
- Vallee M, et al. Neurosteroids: deficient cognitive performance in aged rats depends on low pregnenolone sulfate levels in the hippocampus. Proc Natl Acad Sci USA. 1997;94:14865. [PMC free article: PMC25129] [PubMed: 9405705]
- 204.
- Mayo W, et al. Pregnenolone sulfate enhances neurogenesis and PSA-NCAM in young and aged hippocampus. Neurobiol Aging. 2005;26:103. [PubMed: 15585350]
- 205.
- Sliwinski A, et al. Pregnenolone sulfate enhances long-term potentiation in CA1 in rat hippocampus slices through the modulation of N-methyl-D-aspartate receptors. J Neurosci Res. 2004;78:691. [PubMed: 15505794]
- 206.
- Lynch G, Gall CM. Ampakines and the three-fold path to cognitive enhancement. Trends Neurosci. 2006;29:554. [PubMed: 16890999]
- Review The chemical biology of clinically tolerated NMDA receptor antagonists.[J Neurochem. 2006]Review The chemical biology of clinically tolerated NMDA receptor antagonists.Chen HS, Lipton SA. J Neurochem. 2006 Jun; 97(6):1611-26.
- Review Biology of the NMDA Receptor[ 2009]Review Biology of the NMDA ReceptorVan Dongen AM. 2009
- Review NMDA Receptors in Drosophila.[Biology of the NMDA Receptor. ...]Review NMDA Receptors in Drosophila.Xia S, Chiang AS. Biology of the NMDA Receptor. 2009
- Review Inflammatory mediators leading to protein misfolding and uncompetitive/fast off-rate drug therapy for neurodegenerative disorders.[Int Rev Neurobiol. 2007]Review Inflammatory mediators leading to protein misfolding and uncompetitive/fast off-rate drug therapy for neurodegenerative disorders.Lipton SA, Gu Z, Nakamura T. Int Rev Neurobiol. 2007; 82:1-27.
- Review NMDARs in neurological diseases: a potential therapeutic target.[Int J Neurosci. 2015]Review NMDARs in neurological diseases: a potential therapeutic target.Gonzalez J, Jurado-Coronel JC, Ávila MF, Sabogal A, Capani F, Barreto GE. Int J Neurosci. 2015 May; 125(5):315-27. Epub 2014 Jul 30.
- Activation Mechanisms of the NMDA Receptor - Biology of the NMDA ReceptorActivation Mechanisms of the NMDA Receptor - Biology of the NMDA Receptor
Your browsing activity is empty.
Activity recording is turned off.
See more...