

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
McEntyre J, Ostell J, editors. The NCBI Handbook [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2002-.

This publication is provided for historical reference only and the information may be out of date.

Summary
The comparison of nucleotide or protein sequences from the same or different organisms is a very powerful tool in molecular biology. By finding similarities between sequences, scientists can infer the function of newly sequenced genes, predict new members of gene families, and explore evolutionary relationships. Now that whole genomes are being sequenced, sequence similarity searching can be used to predict the location and function of protein-coding and transcription-regulation regions in genomic DNA.
Basic Local Alignment Search Tool (BLAST) (1, 2) is the tool most frequently used for calculating sequence similarity. BLAST comes in variations for use with different query sequences against different databases. All BLAST applications, as well as information on which BLAST program to use and other help documentation, are listed on the BLAST homepage. This chapter will focus more on how BLAST works, its output, and how both the output and program itself can be further manipulated or customized, rather than on how to use BLAST or interpret BLAST results.
Introduction
The way most people use BLAST is to input a nucleotide or protein sequence as a query against all (or a subset of) the public sequence databases, pasting the sequence into the textbox on one of the BLAST Web pages. This sends the query over the Internet, the search is performed on the NCBI databases and servers, and the results are posted back to the person's browser in the chosen display format. However, many biotech companies, genome scientists, and bioinformatics personnel may want to use “stand-alone” BLAST to query their own, local databases or want to customize BLAST in some way to make it better suit their needs. Stand-alone BLAST comes in two forms: the executables that can be run from the command line; or the Standalone WWW BLAST Server, which allows users to set up their own in-house versions of the BLAST Web pages.
There are many different variations of BLAST available to use for different sequence comparisons, e.g., a DNA query to a DNA database, a protein query to a protein database, and a DNA query, translated in all six reading frames, to a protein sequence database. Other adaptations of BLAST, such as PSI-BLAST (for iterative protein sequence similarity searches using a position-specific score matrix) and RPS-BLAST (for searching for protein domains in the Conserved Domains Database, Chapter 3) perform comparisons against sequence profiles.
This chapter will first describe the BLAST architecture—how it works at the NCBI site—and then go on to describe the various BLAST outputs. The best known of these outputs is the default display from BLAST Web pages, the so-called “traditional report”. As well as obtaining BLAST results in the traditional report, results can also be delivered in structured output, such as a hit table (see below), XML, or ASN.1. The optimal choice of output format depends upon the application. The final part of the chapter discusses stand-alone BLAST and describes possibilities for customization. There are many interfaces to BLAST that are often not exploited by users but can lead to more efficient and robust applications.
How BLAST Works: The Basics
The BLAST algorithm is a heuristic program, which means that it relies on some smart shortcuts to perform the search faster. BLAST performs "local" alignments. Most proteins are modular in nature, with functional domains often being repeated within the same protein as well as across different proteins from different species. The BLAST algorithm is tuned to find these domains or shorter stretches of sequence similarity. The local alignment approach also means that a mRNA can be aligned with a piece of genomic DNA, as is frequently required in genome assembly and analysis. If instead BLAST started out by attempting to align two sequences over their entire lengths (known as a global alignment), fewer similarities would be detected, especially with respect to domains and motifs.
When a query is submitted via one of the BLAST Web pages, the sequence, plus any other input information such as the database to be searched, word size, expect value, and so on, are fed to the algorithm on the BLAST server. BLAST works by first making a look-up table of all the “words” (short subsequences, which for proteins the default is three letters) and “neighboring words”, i.e., similar words in the query sequence. The sequence database is then scanned for these “hot spots”. When a match is identified, it is used to initiate gap-free and gapped extensions of the “word”.
BLAST does not search GenBank flatfiles (or any subset of GenBank flatfiles) directly. Rather, sequences are made into BLAST databases. Each entry is split, and two files are formed, one containing just the header information and one containing just the sequence information. These are the data that the algorithm uses. If BLAST is to be run in “stand-alone” mode, the data file could consist of local, private data, downloaded NCBI BLAST databases, or a combination of the two.
After the algorithm has looked up all possible "words" from the query sequence and extended them maximally, it assembles the best alignment for each query–sequence pair and writes this information to an SeqAlign data structure (in ASN.1 ; also used by Sequin, see Chapter 12). The SeqAlign structure in itself does not contain the sequence information; rather, it refers to the sequences in the BLAST database (Figure 1).
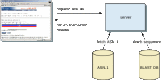
The BLAST Formatter, which sits on the BLAST server, can use the information in the SeqAlign to retrieve the similar sequences found and display them in a variety of ways. Thus, once a query has been completed, the results can be reformatted without having to re-execute the search. This is possible because of the QBLAST system.
BLAST Scores and Statistics
Once BLAST has found a similar sequence to the query in the database, it is helpful to have some idea of whether the alignment is “good” and whether it portrays a possible biological relationship, or whether the similarity observed is attributable to chance alone. BLAST uses statistical theory to produce a bit score and expect value (E-value) for each alignment pair (query to hit).
The bit score gives an indication of how good the alignment is; the higher the score, the better the alignment. In general terms, this score is calculated from a formula that takes into account the alignment of similar or identical residues, as well as any gaps introduced to align the sequences. A key element in this calculation is the “substitution matrix ”, which assigns a score for aligning any possible pair of residues. The BLOSUM62 matrix is the default for most BLAST programs, the exceptions being blastn and MegaBLAST (programs that perform nucleotide–nucleotide comparisons and hence do not use protein-specific matrices). Bit scores are normalized, which means that the bit scores from different alignments can be compared, even if different scoring matrices have been used.
The E-value gives an indication of the statistical significance of a given pairwise alignment and reflects the size of the database and the scoring system used. The lower the E-value, the more significant the hit. A sequence alignment that has an E-value of 0.05 means that this similarity has a 5 in 100 (1 in 20) chance of occurring by chance alone. Although a statistician might consider this to be significant, it still may not represent a biologically meaningful result, and analysis of the alignments (see below) is required to determine “biological” significance.
BLAST Output: 1. The Traditional Report
Most BLAST users are familiar with the so-called “traditional” BLAST report. The report consists of three major sections: (1) the header, which contains information about the query sequence, the database searched (Figure 2). On the Web, there is also a graphical overview (Figure 3); (2) the one-line descriptions of each database sequence found to match the query sequence; these provide a quick overview for browsing (Figure 4); (3) the alignments for each database sequence matched (Figure 5) (there may be more than one alignment for a database sequence it matches).
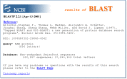


The traditional report is really designed for human readability, as opposed to being parsed by a program. For example, the one-line descriptions are useful for people to get a quick overview of their search results, but they are rarely complete descriptors because of limited space. Also, for convenience, there are several pieces of information that are displayed in both the one-line descriptions and alignments (for example, the E-values, scores, and descriptions); therefore, the person viewing the search output does not need to move back and forth between sections.
New features may be added to the report, e.g., the addition of links to Entrez Gene records (Chapter 19) from sequence hits, which result in a change of output format. These are easy for people to pick up on and take advantage of but can trip programs that parse this BLAST output.
By default, a maximum of 500 sequence matches are displayed, which can be changed on the advanced BLAST page with the Alignments option. Many components of the BLAST results display via the Internet and are hyperlinked to the same information at different places in the page, to additional information including help documentation, and to the Entrez sequence records of matched sequences. These records provide more information about the sequence, including links to relevant research abstracts in PubMed.
BLAST Output: 2. The Hit Table
Although the traditional report is ideal for investigating the characteristics of one gene or protein, often scientists want to make a large number of BLAST runs for a specialized purpose and need only a subset of the information contained in the traditional BLAST report. Furthermore, in cases where the BLAST output will be processed further, it can be unreliable to parse the traditional report. The traditional report is merely a display format with no formal structure or rules, and improvements may be made at any time, changing the underlying HTML. The hit table format provides a simple and clean alternative (Figure 6).

The screening of many newly sequenced human Expressed Sequence Tags (ESTs) for contamination by the Escherichia coli cloning vector is a good example of when it is preferable to use the hit table output over the traditional report. In this case, a strict, high E-value threshold would be applied to differentiate between contaminating E. coli sequence and the human sequence. Those human ESTs that find very strong, near-exact E.coli sequence matches can be discarded without further examination. (Borderline cases may require further examination by a scientist.)
For these purposes, the hit table output is more useful than the traditional report; it contains only the information required in a more formal structure. The hit table output contains no sequences or definition lines, but for each sequence matched, it lists the sequence identifier, the start and stop points for stretches of sequence similarity (offset by one residue), the percent identity of the match, and the E-value.
BLAST Output: 3. Structured Output
There are drawbacks to parsing both the BLAST report and even the simpler hit table. There is no way to automatically check for truncated or otherwise corrupted output in cases when a large number of sequences are being screened. (This may happen if the disk is full, for example.) Also, there is no rigorous check for syntax changes in the output, such as the addition of new features, which can lead to erroneous parsing. Structured output allows for automatic and rigorous checks for syntax errors and changes. Both XML and ASN.1 are examples of structured output in which there are built-in checks for correct and complete syntax and structure. (In the case of XML, for example, this is ensured by the necessity for matching tags and the DTD.) For text reports, there is often no specification, but perhaps a (incomplete) description of the file is written afterward.
ASN.1 Is Used by the BLAST Server
As well as the hit table and traditional report shown in HTML, BLAST results can also be formatted in plain text, XML, and ASN.1 (Figure 7), and what's more, the format for a given BLAST result can be changed without re-executing the search.
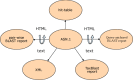
Figure 7
The different output formats that can be produced from ASN.1. Note that some nodes can be viewed as both HTML and text. XML is also structured output but can be produced from ASN.1 because it has equivalent information.
A change in BLAST format without re-executing the search is possible because when a scientist looks at a Web page of BLAST results at NCBI, the HTML that makes that page has been created from ASN.1 (Figure 7). Although the formatted results are requested from the server, the information about the alignments is fetched from a disk in ASN.1, as are the corresponding sequences from the BLAST databases (see Figure 1). The formatter on the BLAST server then puts these results together as a BLAST report. The BLAST search itself has been uncoupled from the way the result is formatted, thus allowing different output formats from the same search. The strict internal validation of ASN.1 ensures that these output formats can always be produced reliably.
Information about the Alignment Is Contained within a SeqAlign
SeqAlign is the ASN.1 object that contains the alignment information about the BLAST search. The SeqAlign does not contain the actual sequence that was found in the match but does contain the start, stop, and gap information, as well as scores, E-values, sequence identifiers, and (DNA) strand information.
As mentioned above, the actual database sequences are fetched from the BLAST databases when needed. This means that an identifier must uniquely identify a sequence in the database. Furthermore, the query sequence cannot have the same identifier as any sequence in the database unless the query sequence itself is in the database. If one is using stand-alone BLAST with a custom database, it is possible to specify that every sequence is uniquely identified by using the –O option with formatdb (the program that converts FASTA files to BLAST database format). This also indexes the entries by identifier. Similarly, the –J option in the (stand-alone) programs blastall, blastpgp, megablast, or rpsblast certifies that the query does not use an identifier already in the database for a different sequence. If the –O and –J options are not used, BLAST assigns unique identifiers (for that run) to all sequences and shields the user from this knowledge.
Any BLAST database or FASTA file from the NCBI Web site that contains gi numbers already satisfies the uniqueness criterion. Unique identifiers are normally a problem only when custom databases are produced and care is not taken in assigning identifiers. The identifier for a FASTA entry is the first token (meaning the letters up to the first space) after the > sign on the definition line. The simplest case is to simply have a unique token (e.g., 1, 2, and so on), but it is possible to construct more complicated identifiers that might, for example, describe the data source. For the FASTA identifiers to be reliably parsed, it is necessary for them to follow a specific syntax (see Appendix 1).
More information on the SeqAlign produced by BLAST can be found here or be downloaded as a PowerPoint presentation, as well as from the NCBI Toolkit Software Developer's handbook.
XML
XML and ASN.1 are both structured languages and can express the same information; therefore, it is possible to produce a SeqAlign in XML. Some users do not find the format of the information in the SeqAlign to be convenient because it does not contain actual sequence information, and when the sequence is fetched from the BLAST database, it is packed two or four bases per byte. Typically, these users are familiar with the BLAST report and want something similar but in a format that can be parsed reliably. The XML produced by BLAST meets this need, containing the query and database sequences, sequence definition lines, the start and stop points of the alignments (one offset), as well as scores, E-values, and percent identity. There is a public DTD for this XML output.
BLAST Code
The BLAST code is part of the NCBI Toolkit, which has many low-level functions to make it platform independent; the Toolkit is supported under Linux and many varieties of UNIX, NT, and MacOS. To use the Toolkit, developers should write a function “Main”, which is called by the Toolkit “main”. The BLAST code is contained mostly in the tools directory (see Appendix 2 for an example).
The BLAST code has a modular design. For example, the Application Programming Interface (API) for retrieval from the BLAST databases is independent of the compute engine. The compute engine is independent from the formatter; therefore, it is possible (as mentioned above) to compute results once but view them in many different modes.
Readdb API
The readdb API can be used to easily extract information from the BLAST databases. Among the data available are the date the database was produced, the title, the number of letters, number of sequences, and the longest sequence. Also available are the sequence and description of any entry. The latest version of the BLAST databases also contains a taxid (an integer specifying some node of the NCBI taxonomy tree; see Chapter 4). Users are strongly encouraged to use the readdb API rather than reading the files associated with the database, because the the files are subject to change. The API, on the other hand, will support the newest version, and an attempt will be made to support older versions. See Appendix 2 for an example of a simple program (db2fasta.c) that demonstrates the use of the readdb API.
Performing a BLAST Search with C Function Calls
Only a few function calls are needed to perform a BLAST search. Appendix 3 shows an excerpt from a Demonstration Program doblast.c.
Formatting a SeqAlign
MySeqAlignPrint (called in the example in Appendix 3) is a simple function to print a view of a SeqAlign (see Appendix 4).
Appendix 1. FASTA identifiers
The syntax of the FASTA definition lines used in the NCBI BLAST databases depends upon the database from which each sequence was obtained (see Chapter 1 on GenBank). Table 1 shows how the sequence source databases are identified.
Table 1
Database identifiers in FASTA definition lines.
For example, if the identifier of a sequence in a BLAST result is gb|M73307|AGMA13GT, the gb tag indicates that sequence is from GenBank, M73307 is the GenBank Accession number, and AGMA13GT is the GenBank locus.
The bar (|) separates different fields. In some cases, a field is left empty, although the original specification called for including this field. To make these identifiers backwards-compatible for older parsers, the empty field is denoted by an additional bar (||).
A gi identifier has been assigned to each sequence in NCBI's sequence databases. If the sequence is from an NCBI database, then the gi number appears at the beginning of the identifier in a traditional report. For example, gi|16760827|ref|NP_456444.1 indicates an NCBI reference sequence with the gi number 16760827 and Accession number NP_456444.1. (In stand-alone BLAST, or when running BLAST from the command line, the –I option should be used to display the gi number.)
The reason for adding the gi identifier is to provide a uniform, stable naming convention. If a nucleotide or protein sequence changes (for example, if it is edited by the original submitter of the sequence), a new gi identifier is assigned, but the Accession number of the record remains unchanged. Thus, the gi identifier provides a mechanism for identifying the exact sequence that was used or retrieved in a given search. This is also useful when creating crosslinks between different Entrez databases (Chapter 15).
Appendix 2. Readdb API
A simple program (db2fasta.c) that demonstrates the use of the readdb API.
Int2 Main (void)
{
BioseqPtr bsp;
Boolean is_prot;
ReadDBFILEPtr rdfp;
FILE *fp;
Int4 index;
if (! GetArgs ("db2fasta", NUMARG, myargs))
{
return (1);
}
if (myargs[1].intvalue)
is_prot = TRUE;
else
is_prot = FALSE;
fp = FileOpen("stdout", "w");
rdfp = readdb_new(myargs[0].strvalue, is_prot);
index = readdb_acc2fasta(rdfp, myargs[2].strvalue);
bsp = readdb_get_bioseq(rdfp, index);
BioseqRawToFasta(bsp, fp, !is_prot);
bsp = BioseqFree(bsp);
rdfp = readdb_destruct(rdfp);
return 0;
}
Note that:
- 1.
Readdb_new allocates an object for reading the database.
- 2.
Readdb_acc2fasta fetches the ordinal number (zero offset) of the record given a FASTA identifier (e.g., gb|AAH06776.1|AAH0676).
- 3.
Readdb_get_bioseq fetches the BioseqPtr (which contains the sequence, description, and identifiers) for this record.
- 4.
BioseqRawToFasta dumps the sequence as FASTA.
Note also that Main is called, rather than “main”, and a call to GetArgs is used to get the command-line arguments. db2fasta.c is contained in the tar archive ftp://ftp.ncbi.nih.gov/blast/demo/blast_demo.tar.gz.
Appendix 3. Excerpt from a demonstration program doblast.c
/* Get default options. */
options = BLASTOptionNew(blast_program, TRUE);
if (options == NULL)
return 5;
options->expect_value = (Nlm_FloatHi) myargs [3].floatvalue;
/* Perform the actual search. */
seqalign = BioseqBlastEngine(query_bsp, blast_program, blast_database, options,
NULL, NULL, NULL);
/* Do something with the SeqAlign... */
MySeqAlignPrint(seqalign, outfp);
/* clean up. */
seqalign = SeqAlignSetFree(seqalign);
options = BLASTOptionDelete(options);
sep = SeqEntryFree(sep);
FileClose(infp);
FileClose(outfp);
The main steps here are:
- 1.
BLASTOptionNew allocates a BLASTOptionBlk with default values for the specified program (e.g., blastp); the Boolean argument specifies a gapped search.
- 2.
The expect_value member of the BLASTOptionBlk is changed to a non-default value specified on the command-line.
- 3.
BioseqBlastEngine performs the search of the BioseqPtr (query_bsp). The BioseqPtr could have been obtained from the BLAST databases, Entrez, or from FASTA using the function call FastaToSeqEntry.
The BLASTOptionBlk structure contains a large number of members. The most useful ones and a brief description for each are listed in Table 2.
Table 2
The most frequently used BLAST options in the BLASTOptionBlk structure.
Appendix 4. A function to print a view of a SeqAlign: MySeqAlignPrint
#define BUFFER_LEN 50
/*
Print a report on hits with start/stop. Zero-offset is used.
*/
static void MySeqAlignPrint(SeqAlignPtr seqalign, FILE *outfp)
{
Char query_id_buf[BUFFER_LEN+1], target_id_buf[BUFFER_LEN+1];
SeqIdPtr query_id, target_id;
while (seqalign)
{
query_id = SeqAlignId(seqalign, 0);
SeqIdWrite(query_id, query_id_buf, PRINTID_FASTA_LONG, BUFFER_LEN);
target_id = SeqAlignId(seqalign, 1);
SeqIdWrite(target_id, target_id_buf, PRINTID_FASTA_LONG, BUFFER_LEN);
fprintf(outfp, "%s:%ld-%ld\t%s:%ld-%ld\n",
query_id_buf, (long) SeqAlignStart(seqalign, 0), (long) SeqAlignStop(seqalign, 0),
target_id_buf, (long) SeqAlignStart(seqalign, 1), (long) SeqAlignStop(seqalign, 1));
seqalign = seqalign->next;
}
return;
}
Note that:
- 1.
SeqAlignId gets the sequence identifier for the zero-th identifier (zero offset). This is actually a C structure.
- 2.
SeqIdWrite formats the information in query_id into a FASTA identifier (e.g., gi|129295) and places it into query_buf.
- 3.
SeqAlignStart and SeqAlignStop return the start values of the zero-th and first sequences (or first and second).
All of this is done by high-level function calls, and it is not necessary to write low-level function calls to parse the ASN.1.
References
- 1.
- Altschul SF , Gish W , Miller W , Myers EW , Lipman DJ . Basic Local Alignment Search Tool. J Mol Biol. 1990;215:403–410. [PubMed: 2231712]
- 2.
- Altschul SF , Madden TL , Schaffer AA , Zhang J , Zhang Z , Miller W , Lipman DJ . Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. [PMC free article: PMC146917] [PubMed: 9254694]
- Summary
- Introduction
- How BLAST Works: The Basics
- BLAST Scores and Statistics
- BLAST Output: 1. The Traditional Report
- BLAST Output: 2. The Hit Table
- BLAST Output: 3. Structured Output
- BLAST Code
- Appendix 1. FASTA identifiers
- Appendix 2. Readdb API
- Appendix 3. Excerpt from a demonstration program doblast.c
- Appendix 4. A function to print a view of a SeqAlign: MySeqAlignPrint
- References
- The BLAST Sequence Analysis Tool - The NCBI HandbookThe BLAST Sequence Analysis Tool - The NCBI Handbook
Your browsing activity is empty.
Activity recording is turned off.
See more...