Clinical characteristics.
Retinoblastoma is a malignant tumor of the developing retina that occurs in children, usually before age five years. Retinoblastoma may be unifocal or multifocal. About 60% of affected individuals have unilateral retinoblastoma with a mean age of diagnosis of 24 months; about 40% have bilateral retinoblastoma with a mean age of diagnosis of 15 months. Heritable retinoblastoma is associated with susceptibility for retinoblastoma as well as non-ocular tumors.
Diagnosis/testing.
The diagnosis of retinoblastoma is usually established by examination of the fundus of the eye using indirect ophthalmoscopy. Imaging studies can be used to support the diagnosis and stage the tumor. Almost all retinoblastomas develop following biallelic inactivation of RB1 in a cone cell precursor in the developing retina. The diagnosis of heritable retinoblastoma is established in a proband with retinoblastoma or retinoma (benign potential precursor to retinoblastoma) and a family history of retinoblastoma or a heterozygous germline pathogenic variant in RB1 identified by molecular genetic testing. Epigenetic hypermethylation of the RB1 promotor also results in inactivation of RB1 but cannot be transmitted via the germline. Amplification of MYCN in a developing retinal cone cell can also result in retinoblastoma that is not thought to be heritable.
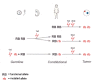
The following staging has been recommended for individuals with retinoblastoma and/or risk of heritable retinoblastoma to describe the genetic risk of a germline pathogenic variant in RB1:
HX. Individual with unknown or insufficient evidence of a constitutional (germline) RB1 pathogenic variant
H0. Individual who did not inherit a known familial germline RB1 pathogenic variant confirmed by molecular genetic testing
(H0*. Individual with unilateral retinoblastoma or retinoma with no germline RB1 pathogenic variant identified on molecular genetic testing; residual risk of mosaicism is <1%.)
H1. Individual with bilateral retinoblastoma, trilateral retinoblastoma (retinoblastoma with intracranial central nervous system midline embryonic tumor), retinoblastoma and a family history of retinoblastoma, or identification of a germline RB1 pathogenic variant
Management.
Treatment of manifestations: Early diagnosis and treatment of retinoblastoma can reduce morbidity and increase longevity; care is best provided by multidisciplinary teams of specialists including ophthalmology, pediatric oncology, pathology, and radiation oncology. Treatment options depend on tumor stage, number of tumor foci (unifocal, unilateral multifocal, or bilateral), localization and size of the tumor(s) within the eye(s), presence of vitreous seeding, potential for useful vision, extent and kind of extraocular extension, and resources available. Treatment options include enucleation; cryotherapy; laser; systemic or local ocular chemotherapy, including intra-arterial chemotherapy, combined with or followed by laser or cryotherapy; radiation therapy using episcleral plaques; and, as a last resort, external beam radiotherapy. Standard treatments for non-ocular neoplasms.
Surveillance: Individuals with an RB1 germline pathogenic variant (H1) should have eye examinations (under anesthesia in young children) every three to four weeks until age six months, every two months until age three years, every three to six months until age seven years, annually until age ten years, and then every two years in order to identify a retinoblastoma as early as possible. In individuals with unilateral retinoblastoma without an identified heterozygous germline RB1 pathogenic variant (H0*), clinical eye examination and eye ultrasound every three to six months until age seven years, then every two years. In individuals with retinomas, retinal examinations and imaging every one to two years. To detect non-ocular tumors, prompt clinical investigation of any signs and/or symptoms of malignant neoplasms.
Agents/circumstances to avoid: If possible, radiation (including x-rays, CT scans, and external beam radiation) and DNA damaging agents (tobacco, UV light) should be avoided in individuals with heritable retinoblastoma (H1) to minimize the lifetime risk of developing subsequent malignant neoplasms. It is plausible that cancer risks in these individuals may also be reduced by limiting exposure to chemotherapy.
Evaluation of relatives at risk: Molecular genetic testing if the pathogenic variant in the family is known (which can reduce the need for costly screening procedures in those family members who have not inherited the pathogenic variant [i.e., H0]) or eye examinations by an ophthalmologist experienced in the treatment of retinoblastoma if the pathogenic variant is not known for early identification of asymptomatic at-risk children.
Genetic counseling.
Heritable retinoblastoma is inherited in an autosomal dominant manner. The majority of individuals with heritable retinoblastoma represent simplex cases (i.e., the only person in the family known to be affected). Some individuals diagnosed with heritable retinoblastoma inherited an RB1 pathogenic variant from an affected parent. Each child of an individual with heritable retinoblastoma (H1) has a 50% chance of inheriting the RB1 pathogenic variant. If the RB1 pathogenic variants that have been detected in tumor tissue are not detected in DNA from leukocytes of the proband, there is an estimated 1.2% chance that the proband has germline mosaicism for one of the pathogenic variants identified in the tumor tissue and the offspring of the proband are at a 0.6% risk of inheriting a germline pathogenic variant. If one of the pathogenic variants identified in the proband's tumor is found to be mosaic in DNA from leukocytes of the proband, the level of germline involvement is uncertain. Predictive DNA testing in offspring is possible if the cancer-predisposing germline RB1 variant has been identified in the proband. If molecular genetic testing is not available or is uninformative in the proband, empiric risks to offspring based on tumor presentation and family history can be used. If the germline RB1 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.