Clinical Description
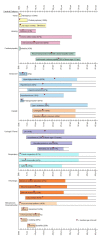
The first clinical manifestation of Alström syndrome (Table 3) is usually nystagmus caused by cone-rod dystrophy and/or infantile-onset cardiomyopathy. Later-onset findings include obesity that manifests during the first years of life, progressive sensorineural hearing loss (SNHL), insulin resistance / T2DM, adolescent- or adult-onset restrictive cardiomyopathy, hepatic steatosis, and progressive renal dysfunction. Wide clinical variability is observed among individuals with Alström syndrome, including among sibs [Hoffman et al 2005].
Table 3.
Age of Onset and Incidence of Common Features of Alström Syndrome
View in own window
| Feature | Age of Onset Range (Mean) | Incidence 1 |
|---|
| Cone-rod dystrophy | Birth - 15 mos (5 mos) 2 | 100% |
| Obesity | Birth - 5 years (2.5 yrs) | 98% |
| Progressive SNHL | 2-25 yrs (9 yrs) | 88% |
| Cardiomyopathy | Infantile | 2 wks - 4 mos | 42% 3 |
| Restrictive | Juvenile - late 30s | 18% |
| Insulin resistance / T2DM | 4-30 yrs / 8-40 yrs (16 yrs) | 92% / 68% |
| Short stature | Puberty - adult | 98% |
| Hypogonadism (central or primary) | 10+ yrs | 78% of males |
| Urologic disease (bladder dysfunction) | Adolescence - adult | 48% |
| Progressive renal disease | Adolescence - adult | Variably progressive in all individuals |
| Hepatic disease | 8-30 yrs | 23%-100% |
SNHL = sensorineural hearing loss; T2DM = type 2 diabetes mellitus
- 1.
Given the age-dependent nature of many features of Alström syndrome, percentages are not exact and may be underestimates.
- 2.
Rare individuals with atypical later onset and milder retinal dystrophy are reported.
- 3.
The proportion of infants with Alström syndrome who develop infantile-onset cardiomyopathy is probably underestimated because some infants succumb to heart failure before the diagnosis of Alström syndrome is made.
Cone-rod dystrophy. In most affected individuals, visual problems present as progressive cone dystrophy resulting in visual impairment, photophobia, and nystagmus between birth and age 15 months. Rod function is preserved initially but deteriorates as the individual ages, with visual acuity of 6/60 or less by age ten years, increasing constriction of visual fields, and no light perception by age 35 years [Nasser et al 2018].
Optical coherence tomography (OCT) reveals central macular changes that are mild during early childhood but slowly progress, resulting in loss of photoreceptors and retinal pigment epithelium. Severe retinal wrinkling, intraretinal opacities, foveal contour abnormalities, optic nerve drusen, vitreoretinal separation, and hyperreflectivities in all retinal layers are observed. The severity of the macular changes on OCT correlates with vision [Dotan et al 2017]. The severity and age of onset of the retinal degeneration vary among affected individuals [Malm et al 2008]. While many individuals lose all perception of light by the end of the second decade, a minority retain the ability to read large print into the third decade. Posterior subcapsular cataracts are common, even in the absence of diabetes.
Obesity. Children with Alström syndrome have normal birth weight. Hyperphagia and excessive weight gain begin during the first years, resulting in childhood obesity. In some individuals body weight tends to normalize, decreasing into the high-normal to normal range after adolescence.
Progressive bilateral SNHL presents in the first decade in as many as 70% of individuals with Alström syndrome; average age at detection of hearing loss is seven years [Marshall et al 2005, Ozantürk et al 2015, Lindsey et al 2017]. A majority of affected infants pass newborn screening for hearing loss [Lindsey et al 2017]. Hearing loss may be detected as early as age one year. Initially in the high frequency range, hearing loss may progress to the severe or moderately severe range (40-70 db) by the end of the first to second decade [Van den Abeele et al 2001].
In 33 individuals with Alström syndrome, the average rate of progression of hearing loss was 10-15 db/decade [Lindsey et al 2017]. The auditory defect in Alström syndrome is mapped to the outer hair cells of cochlea based on absent otoacoustic emissions, intact speech discrimination, and disproportionately normal auditory brain stem responses [Lindsey et al 2017]. Therefore, individuals with Alström syndrome are good candidates for aural amplification or cochlear implantation in cases of severe-to-profound hearing loss [Florentzson et al 2010, Lindsey et al 2017].
A high incidence of adhesive otitis media (glue ear) due to long-standing fluid in the middle ear can lead to an additional conductive hearing loss [Marshall et al 2005].
Cardiomyopathy. More than 60% of individuals with Alström syndrome develop congestive heart failure at some stage of their lives as a result of infantile-, adolescent-, or adult-onset cardiomyopathy. Onset, progression, and clinical outcome of cardiomyopathy can vary, even within families [Hoffman et al 2005, Mahamid et al 2013, Brofferio et al 2017].
More than 40% of infants with Alström syndrome present with a transient but severe cardiomyopathy with onset between ages three weeks and four months [Marshall et al 2005, Brofferio et al 2017]. Of note, the proportion of those with Alström syndrome who develop infantile-onset cardiomyopathy may be underestimated because some infants may succumb before the diagnosis of Alström syndrome [Louw et al 2014]. Most infants with severe cardiomyopathy develop irreversible heart failure, leading to death within the first weeks to months of life. Autopsy findings in these neonates show dramatically increased mitotic activity in the cardiomyocytes (i.e., mitogenic cardiomyopathy) [Louw et al 2014, Shenje et al 2014]. Most infants who survive make an apparently full recovery by age two years.
About 20% of individuals with Alström syndrome develop a later-onset progressive restrictive cardiomyopathy identified between the teens and late 30s. A characteristic feature of these individuals appears to be myocardial fibrosis documented at postmortem [Marshall et al 2005] and on cardiovascular MRI in asymptomatic and clinically affected individuals [Loudon et al 2009, Corbetti et al 2013, Edwards et al 2015]. Strain echocardiography suggests that some degree of myocardial fibrosis is probably present in almost all individuals with Alström syndrome including asymptomatic children [Brofferio et al 2017].
Flow-limiting coronary artery disease occurs in approximately 10% but does not appear to be related to progression of myocardial fibrosis.
Severe insulin resistance / type 2 diabetes mellitus (T2DM) are hallmarks of Alström syndrome. The age at which T2DM develops varies; it has been reported at as early as age five years. Insulin resistance proceeds to T2DM in the majority by the third decade. Insulin resistance results in the skin changes of acanthosis nigricans; i.e., velvety hyperpigmented patches in intertriginous areas [Akdeniz et al 2011, Han et al 2018]. An obesity-related contrast in the incidence of T2DM has been described in Canadian versus Italian cohort studies of children with Alström syndrome followed from childhood through to adolescence, with much lower diabetes rates in the leaner Italian children [Mokashi & Cummings 2011, Bettini et al 2012].
In a small study of 12 unrelated individuals with Alström syndrome, obesity (BMI and waist circumference) decreased with age, whereas insulin resistance increased with age [Minton et al 2006]. The hyperinsulinism was out of proportion to the degree of obesity. Consistently, in a recent study comparing 38 individuals with Alström syndrome to 76 age-, sex-, and BMI-matched controls, the severity of insulin resistance in individuals with Alström syndrome was documented to be more than five times that of equally obese controls [Han et al 2018], and metabolic syndrome (defined as ≥3 of the following: abdominal obesity, hypertriglyceridemia, low HDL-cholesterol, hypertension, impaired glucose tolerance) was ten times more common in Alström syndrome compared to controls.
Coronary artery disease as a result of insulin resistance, diabetes, dyslipidemia, and renal failure was reported in one affected individual [Jatti et al 2012]. A more recent cohort study of individuals from the UK has shown that duration of diabetes is linked with increased carotid-femoral pulse wave velocity and that this in turn predicts occurrence of atherosclerosis [Paisey et al 2015]. The authors' unpublished data show coronary artery disease to be increasingly common in adults with Alström syndrome (observed in up to 10%).
Diabetic peripheral neuropathy with risk of foot ulceration occurs rarely if at all in Alström syndrome [Paisey et al 2009] in contrast to patients with adolescent-onset T2DM without Alström syndrome, in whom 30% had severe peripheral neuropathy.
Hyperlipidemia. Insulin resistance is associated with hypertriglyceridemia in >90% of individuals with Alström syndrome [Paisey et al 2004, Paisey et al 2009, Han et al 2018]. Serum triglyceride levels commonly range from 2 to 5 mmol/L (177 to 443 mg/dL) with HDL cholesterol <1 mmol/L (39 mg/dL) and total cholesterol levels from 5 to 7 mmol/L (193 to 271 mg/dL). In some cases serum triglyceride levels can increase to well above 20 mmol/L (1,770 mg/dL). Affected individuals are at risk for sudden increase in triglycerides, precipitating life-threatening pancreatitis [Marshall et al 2005].
Short stature. Growth rates for young children are normal, but accelerated skeletal maturity (2-3 years advanced bone age) and low-serum growth hormone concentrations result in adult stature that is typically below the 25th centile. In about 98% of individuals older than age 16 years height is below the fifth centile [Maffei et al 2007].
Male pubertal development. A variable combination of hypogonadotropic hypogonadism and testicular fibrosis can result in delayed or arrested puberty in males, resulting in normal or immature secondary sexual characteristics; gynecomastia may be present [Marshall et al 2005]. Male fertility, which has not been systematically studied, has not been reported in males with biallelic ALMS1 pathogenic variants.
Female pubertal development. Endocrine disturbances in females include reduced plasma gonadotropin concentrations, hirsutism, polycystic ovarian syndrome associated with insulin resistance, precocious puberty, irregular menses, or amenorrhea. External genitalia are normal in females, though breast development is often poor.
Fertility in females has not been systematically studied. Although a molecular diagnosis was not confirmed, one clinical report describes two unrelated females with late presentation of the syndrome, each of whom had healthy children [Iannello et al 2004].
Urologic disorders of varying severity, which affect approximately 50% of affected individuals, can include detrusor-urethral dyssynergia (lack of coordination of bladder and urethral muscle activity). The greatest problems appear to occur in females in their late teens. Minor manifestations include urgency and long intervals between voiding, suggesting decreased bladder sensation, hesitancy, and poor urinary flow. Moderate manifestations include urinary frequency, incontinence, and symptoms associated with recurrent infections. More severe manifestations are rare (<2%) and include worsening urinary incontinence or retention; these symptoms may alternate. Lower abdominal and perineal pain is common and may relate to abnormal bladder/sphincter function [MacDermott 2001].
Renal disease is common, slowly progressive, and highly variable [Waldman et al 2018]. Onset can be in mid-childhood through adulthood. End-stage renal disease can occur as early as the mid- to late teens.
Renal ultrasonography and MRI may reveal abnormalities [Waldman et al 2018]. The most common ultrasonography finding is renal parenchymal hyperechogenicity often limited to the medulla. Renal cysts are identified in a small number of patients [Waldman et al 2018, Baig et al 2020].
Renal biopsy often shows interstitial fibrosis, glomerular hyalinosis, and tubular atrophy but absence of histopathologic features of diabetic or reflux nephropathy [Marshall et al 2005, Baig et al 2020]. In addition, glomerular function in Alström syndrome does not show significant association with T2DM, hyperlipidemia, cardiomyopathy, or hypertension, suggesting that kidney disease is a primary manifestation of the syndrome. Diabetes and hypertension may have an additive effect on the progression of renal disease.
Obstructive uropathy is rare.
Hepatic disease. Individuals with Alström syndrome have disproportionately high liver fat in comparison to equally obese controls [Han et al 2018]. Their risk of advanced NAFLD and cirrhosis is disproportionately increased for their age, BMI, and duration of diabetes [Gathercole et al 2016]. Plasma concentration of liver enzymes is often elevated starting in early childhood. Hepatomegaly is common. In some affected individuals liver disease progresses to cirrhosis and hepatic failure in the second to third decade. Portal hypertension associated with splenomegaly, esophageal varices, ascites, and hepatic encephalopathy may occur.
Liver biopsies and postmortem examination have revealed varying degrees of steatohepatitis, hepatic fibrosis, cirrhosis, chronic nonspecific active hepatitis with lymphocytic infiltration, and patchy necrosis [Quiros-Tejeira et al 2001, Marshall et al 2005]. Early stages of steatohepatitis can remit and relapse with significant improvements in exercise tolerance, insulin resistance, and blood sugar [Paisey et al 2014].
Gastrointestinal disease. General gastrointestinal disturbances such as epigastric pain and gastroesophageal reflux disease are common.
Pulmonary involvement ranges in severity from frequent upper and lower respiratory infections to pulmonary fibrosis and pulmonary hypertension. Recurrent upper and lower respiratory infections are common at all ages. Evaluation of pulmonary function is problematic because individuals with Alström syndrome have difficulty with deep inspiration / forced expiration. Most frequently there is restrictive lung disease due to kyphoscoliosis, sometimes in combination with pulmonary fibrosis, which has been confirmed in postmortem tissue. This may progress (with the added effects of cardiomyopathy) to pulmonary hypertension. The resulting susceptibility to severe hypoxia postoperatively or during episodes of pneumonia has been reported [Khoo et al 2009, Florentzson et al 2010].
A study of the burden of otosinopulmonary disease in 38 individuals with Alström syndrome revealed that recurrent otitis media was ubiquitous (92%), with 50% requiring pressure-equalization tube placement [Boerwinkle et al 2017]. A history of bronchitis/pneumonia and sinusitis was reported in 61% and 50% of individuals, respectively. However, manifestations of primary ciliary dyskinesia (PCD) (laterality defects, unexplained neonatal respiratory distress, year-round nasal congestion, and wet cough) were far less prevalent in individuals with Alström syndrome compared to those with PCD. In addition, the average nasal nitric oxide production in this cohort was 232 ± 57.1 nL/min compared to <77 nL/min required for a diagnosis of PCD.
Other findings include the following [Marshall et al 2005]:
Severe flat feet (pes planus)
Dental abnormalities
Hypertension, often beginning in childhood (30%)
Delay in early developmental milestones in ~20% of affected individuals, most commonly in gross and fine motor skills; ~30% have a learning disability. Cognitive impairment (IQ <70) is very rare.