

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006.

Baseline data from a cohort of 815 patients (48% males and 52% females) with Fabry disease from 87 centres in 13 European countries who were enrolled in FOS – the Fabry Outcome Survey – were analysed in terms of demography and clinical manifestations of Fabry disease. From this database, it can be seen that there is a significant period of time between the onset of symptoms and diagnosis, with an average delay of 12.4 ± 15.0 years in females and 12.2 ± 13.0 years in males. The database also confirms that heterozygous females should be considered as patients rather than carriers and that children with Fabry disease are symptomatic. Furthermore, data are now available from 483 patients (60% males and 40% females) who are receiving enzyme replacement therapy (ERT), with 224 patients having received ERT for more than 3 years.
FOS – the Fabry Outcome Survey – is a European outcomes database for patients with Fabry disease who are receiving, or are candidates for, enzyme replacement therapy (ERT) with agalsidase alfa. FOS is the world's most comprehensive database of patients with Fabry disease. Data from all consenting patients are entered into the database following a structured clinical assessment by a physician or a nurse specialist. By pooling data from different specialist centres, FOS enables the natural history of this rare disease to be studied in a large group of patients, including men, women and children, and provides baseline data against which the effects of treatment with agalsidase alfa can be measured.
Patient demographics
Since the start of FOS in 2001 there has been a continuous increase in the number of patients who are included in the database (Figure 1). At the end of 2005, data on 815 patients, who were recruited from 87 centres in 13 European countries, had been entered. So far, the largest groups of patients come from Germany (22%) and the UK (15%), whereas most other countries each contribute less than 10% of the patient population.
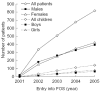
Figure 1
Number of patients with Fabry disease enrolled in FOS – the Fabry Outcome Survey –over time. Data shown include all patients enrolled by the end of each year.
In the first few years of FOS, there were more males than females in the database [1]; however, in the last quarter of 2004, for the first time, the number of heterozygotes exceeded the number of hemizygotes (348 females, 328 males) (Figure 1). At entry into FOS, males were significantly younger than females (p = 0.0045) and they were diagnosed at an earlier age (p < 0.0001) (Table 1). In many cases (35%), the diagnosis of Fabry disease was suspected by other affected family members; however, a range of different medical specialists were involved in diagnosis, including nephrologists (14%), geneticists (10%), paediatricians (8%), dermatologists (7%), general practitioners (5%), cardiologists (5%) and a range of other physicians. Data show that there is no difference in the severity of Fabry disease in patients diagnosed as a result of examinations following the suspicions of relatives or geneticists and those diagnosed by other physicians (FOS-Mainz Severity Score Index, 14.7 versus 15.2, respectively, adjusted for gender and age).
Table 1
Characteristics of 815 patients with Fabry disease at entry into FOS – the Fabry Outcome Survey – (data until end of 2005). Values are expressed as means ± SD.
Delay in diagnosis
Previous studies have reported a significant delay in diagnosis and a wide range of prior misdiagnoses [1, 2]. However, early diagnosis is essential to enable optimal symptomatic management which, in combination with ERT, may improve quality of life and prevent late complications.
Although awareness of the clinical manifestations of Fabry disease has been increased dramatically by publications and scientific presentations, the latest analysis of FOS data shows that there is still a noteworthy time delay between the onset of symptoms and diagnosis (Table 1). The mean time (± SD) between the onset of symptoms and diagnosis was 12.4 ± 15.0 years in females and 12.2 ± 13.0 years in males.
Reported signs and symptoms in male and female patients
The main signs and symptoms in male and female patients, according to age at entry into FOS, are shown in Tables 2 and 3. Multiple organ systems were involved in the majority of patients with Fabry disease. Only 36 of the patients in FOS (5 males, 31 females) had no reported signs and symptoms of Fabry disease at their latest visit. These patients were generally young (males, 20.8 ± 14.1 years; females, 26.0 ± 18.6 years).
Table 2
Frequency and age at onset of specific signs and symptoms of Fabry disease in male patients (n = 375) enrolled in FOS – the Fabry Outcome Survey.
Table 3
Frequency and age at onset of specific signs and symptoms of Fabry disease in female patients (n = 396) enrolled in FOS – the Fabry Outcome Survey.
There is no evidence for an atypical variant of Fabry disease with manifestations limited to a single organ system, as has been reported previously involving the heart [3], kidney [4] and kidney plus heart [5]. In both genders, the number of organ systems involved rises progressively with age, although some signs and symptoms are more common in children than in adults.
Approximately 83% of males and 66% of females had neurological symptoms, the most prevalent of these being neuropathic pain. These symptoms began at an early age, being reported during childhood in males (mean age, 16.3 years) and during early adulthood in women (mean age, 21.2 years). Dermatological manifestations, which were more common in males than in females (70% versus 37%), began at an early age in males (mean age, 18.7 years) and somewhat later in females (mean age, 28.9 years). Other early manifestations of Fabry disease included those involving the gastrointestinal system, which began at a mean age of 23.3 years in males and 25.9 years in females. In total, some 56% of males and 45% of females with Fabry disease reported gastrointestinal problems.
Auditory signs and symptoms, including tinnitus, hearing difficulties or hearing loss, were found in 56% of males and 42% of females. Ocular manifestations were reported in 62.1% of males and nearly 50% of females.
Cardiac and renal manifestations of Fabry disease were reported to begin on average during the fourth decade of life in both men and women in FOS. Cardiac manifestations were reported in 61% of males and 52% of females, beginning at a mean age of 32.9 years in males and 36.4 years in females. Renal/urinary manifestations were reported in 57% of males and 37% of females, with onset at a mean age of 33.9 and 36.8 years, respectively (see Chapter 21). Cerebrovascular events (stroke, transient ischaemic attacks [TIAs] and prolonged reversible ischaemic neurological deficits) were less common, affecting approximately 20% of males and females; however, they may be observed in relatively young patients. The mean age for such events was 33.5 years in males and 41.4 years in females. The youngest patient to report a TIA was a boy aged 12 years. This is the first report of TIA in a child with Fabry disease.
These data confirm that female heterozygotes may exhibit the full range of disease manifestations and should no longer be considered as asymptomatic 'carriers'. However, these data and other clinical studies have shown that the signs and symptoms in females with Fabry disease are more variable than those in males, and that the disease appears to progress more slowly [6, 7]. The manifestations of Fabry disease in females are discussed in more detail in Chapter 30.
Children in FOS
As shown in Figure 1, there has been a steady increase in the number of patients younger than 18 years of age enrolled in FOS. In 2002, 10% of all patients included in FOS were children, but this proportion has increased to 17% in 2005 (see Table 4). Common clinical symptoms reported in children include acroparaesthesiae, decreased sweating and cardiac abnormalities [8] (see Chapter 31).
Mortality
Age and cause of death have been reported by those enrolled in FOS for 68 male and 39 female relatives who were presumed to have Fabry disease. The mean age at death (± SD) of affected male and female relatives was 44.9 ± 9.9 and 57.8 ± 14.3 years, respectively. This significant reduction in life expectancy is consistent with previous studies that have shown a reduced lifespan in males and females with Fabry disease [9, 10].
Enzyme replacement therapy
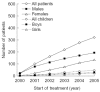
FOS data show, that the number of patients receiving agalsidase alfa has increased progressively over 3 years (Figure 2). In total, 483 patients are currently receiving ERT (60% males). Overall, 73% of all males and 46% of all females in FOS are receiving agalsidase alfa. Of these patients, 67 are children (29 girls and 38 boys). The proportion of women and children receiving ERT is increasing (Table 4). Some 224 patients have now received treatment for more than 3 years. Analysis of data from the latest visit show that those patients who started treatment more recently are generally less severely affected than those who started ERT when it first became available (Figure 3). In 2001, the average age of females who started ERT was about 45 years; by 2005, younger heterozygotes with generally milder disease manifestations were receiving treatment (Figure 4).
![Figure 3. Disease severity (as assessed by FOS –the Fabry Outcome Survey – adaptation of the Mainz Severity Score Index [FOS-MSSI]) at latest visit in males and females (combined) in FOS according to when they began enzyme replacement therapy.](/books/NBK11581/bin/ch16f3.gif)

Figure 4
Age of females in FOS – the Fabry Outcome Survey – when they started enzyme replacement therapy (ERT), according to year.
Conclusions
The FOS database provides detailed information about the clinical manifestations of Fabry disease in a large cohort of patients. The size of this database will soon increase with the inclusion of patients currently enrolled in the US outcomes database for patients treated with agalsidase alfa (FIRE – Fabry International Research Exchange). Through FOS, significant clinical symptoms have been demonstrated in children and heterozygous females. Information gained from analyses of the FOS database may help to increase the awareness of Fabry disease in both genders and to avoid misdiagnoses and delays between the onset of symptoms and diagnosis.
References
- 1.
- Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C. et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004;34:236–42. [PubMed: 15025684]
- 2.
- Galanos J, Nicholls K, Grigg L, Kiers L, Crawford A, Becker G. Clinical features of Fabry's disease in Australian patients. Intern Med J. 2002;32:575–84. [PubMed: 12512750]
- 3.
- Sachdev B, Takenaka T, Teraguchi H, Tei C, Lee P, McKenna WJ. et al. Prevalence of Anderson–Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation. 2002;105:1407–11. [PubMed: 11914245]
- 4.
- Sawada K, Mizoguchi K, Hishida A, Kaneko E, Koide Y, Nishimura K. et al. Point mutation in the α-galactosidase A gene of atypical Fabry disease with only nephropathy. Clin Nephrol. 1996;45:289–94. [PubMed: 8738659]
- 5.
- Germain DP. A new phenotype of Fabry disease with intermediate severity between the classical form and the cardiac variant. Contrib Nephrol. 2001;136:234–40. [PubMed: 11688386]
- 6.
- Whybra C, Kampmann C, Willers I, Davies J, Winchester B, Kriegsmann J. et al. Anderson–Fabry disease: clinical manifestations of disease in female heterozygotes. J Inherit Metab Dis. 2001;24:715–24. [PubMed: 11804208]
- 7.
- Guffon N. Clinical presentation in female patients with Fabry disease. J Med Genet. 2003;40:e38. [PMC free article: PMC1735431] [PubMed: 12676911]
- 8.
- Ries M, Gupta S, Moore DF, Sachdev V, Quirk JM, Murray GJ. et al. Pediatric Fabry disease. Pediatrics. 2005;115:e344–55. [PubMed: 15713906]
- 9.
- MacDermot KD, Holmes A, Miners AH. Anderson–Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet. 2001;38:769–75. [PMC free article: PMC1734754] [PubMed: 11732485]
- 10.
- MacDermot KD, Holmes A, Miners AH. Anderson–Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet. 2001;38:750–60. [PMC free article: PMC1734761] [PubMed: 11694547]
- Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey.[Eur J Clin Invest. 2004]Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey.Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C, Linhart A, Sunder-Plassmann G, Ries M, Beck M. Eur J Clin Invest. 2004 Mar; 34(3):236-42.
- Gender Differences in the Application of Spanish Criteria for Initiation of Enzyme Replacement Therapy for Fabry Disease in the Fabry Outcome Survey.[Int J Mol Sci. 2016]Gender Differences in the Application of Spanish Criteria for Initiation of Enzyme Replacement Therapy for Fabry Disease in the Fabry Outcome Survey.Barba-Romero MÁ, Pintos-Morell G. Int J Mol Sci. 2016 Nov 24; 17(12). Epub 2016 Nov 24.
- Hyperhidrosis: a new and often early symptom in Fabry disease. International experience and data from the Fabry Outcome Survey.[Int J Clin Pract. 2006]Hyperhidrosis: a new and often early symptom in Fabry disease. International experience and data from the Fabry Outcome Survey.Lidove O, Ramaswami U, Jaussaud R, Barbey F, Maisonobe T, Caillaud C, Beck M, Sunder-Plassmann G, Linhart A, Mehta A, et al. Int J Clin Pract. 2006 Sep; 60(9):1053-9.
- Review Fabry disease in females: clinical characteristics and effects of enzyme replacement therapy.[Fabry Disease: Perspectives fr...]Review Fabry disease in females: clinical characteristics and effects of enzyme replacement therapy.Deegan PB, Bähner F, Barba M, Hughes DA, Beck M. Fabry Disease: Perspectives from 5 Years of FOS. 2006
- Review Nervous system manifestations of Fabry disease: data from FOS – the Fabry Outcome Survey.[Fabry Disease: Perspectives fr...]Review Nervous system manifestations of Fabry disease: data from FOS – the Fabry Outcome Survey.Ginsberg L. Fabry Disease: Perspectives from 5 Years of FOS. 2006
- Demographics of FOS – the Fabry Outcome Survey - Fabry DiseaseDemographics of FOS – the Fabry Outcome Survey - Fabry Disease
Your browsing activity is empty.
Activity recording is turned off.
See more...